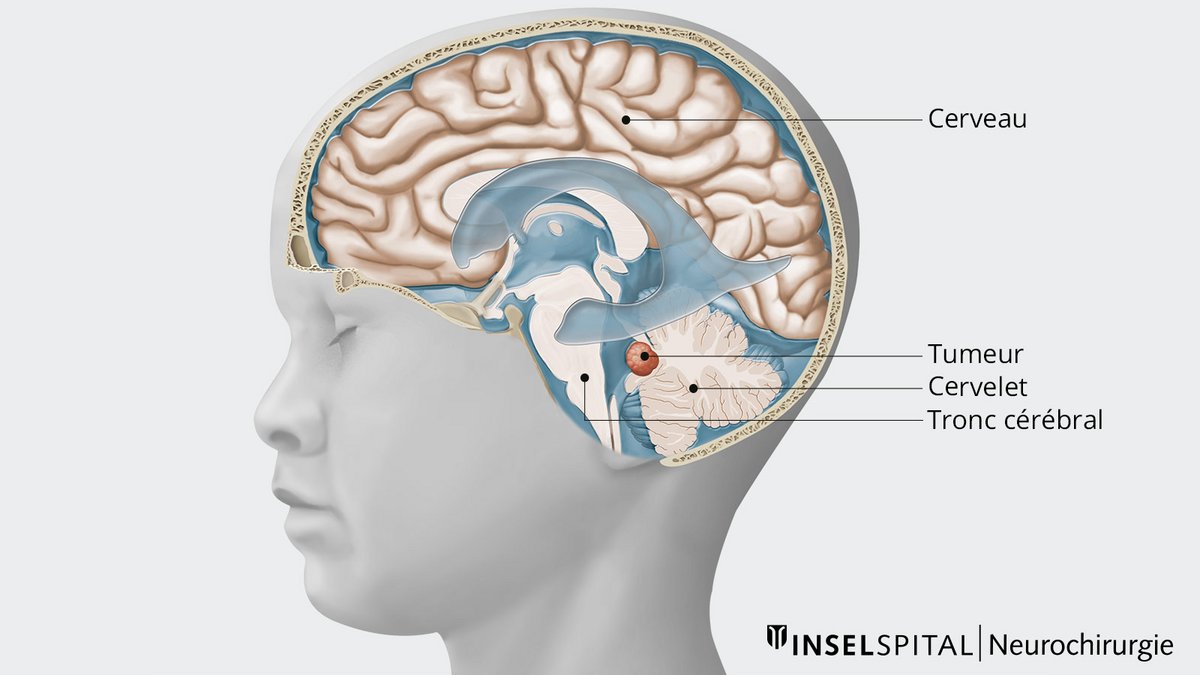
Les médulloblastomes sont les tumeurs cérébrales malignes primaires les plus fréquentes chez l'enfant. Elles sont responsables d'environ 10 % de toutes les tumeurs cérébrales chez les enfants. Rarement, elles apparaissent également chez les adultes, mais elles ne représentent alors que moins de 1 % de toutes les tumeurs cérébrales. La localisation la plus fréquente est au niveau du cervelet. Le traitement standard consiste en une opération suivie, selon l'âge, d'une chimiothérapie et/ou d'une radiothérapie, qui sont adaptées individuellement. Le pronostic et le traitement individuel dépendent en outre fortement du sous-type de tumeur.
Signification clinique
Le médulloblastome est une tumeur embryonnaire, c'est-à-dire qu'il naît de cellules immatures et indifférenciées du système nerveux central (SNC) et se développe très rapidement. Parallèlement, l'analyse génétique des tumeurs a permis d'identifier 4 sous-groupes moléculaires de médulloblastomes, qui diffèrent par leurs caractéristiques génétiques moléculaires et ont également des pronostics différents.
Les médulloblastomes sont typiquement localisés dans la fosse crânienne postérieure, dans la région de la 4e chambre du liquide céphalorachidien (ventricule). La propagation des cellules tumorales par le liquide céphalo-rachidien à d'autres régions du cerveau ou à la moelle épinière est possible. Le plus souvent, le médulloblastome infiltre localement le cervelet et le tronc cérébral. La propagation au cerveau est plus fréquente chez les adultes.
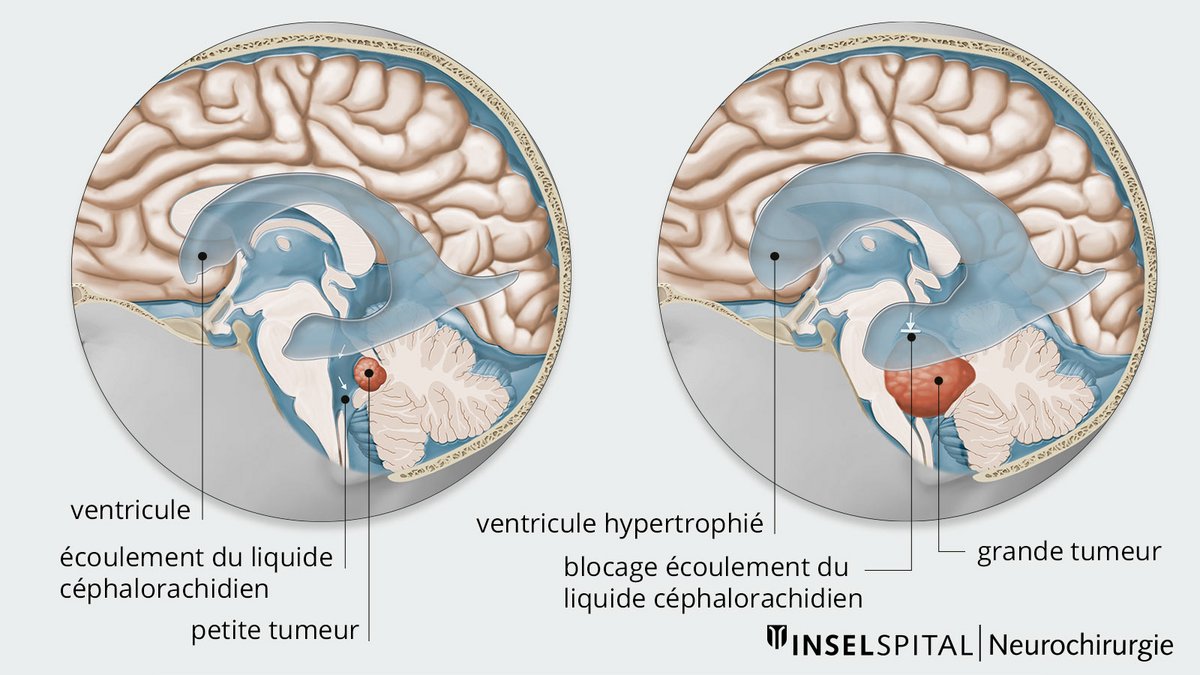
En raison de la croissance rapide et agressive des médulloblastomes, les structures vitales adjacentes peuvent être déplacées et endommagées. En outre, en fonction de sa localisation exacte, la tumeur peut provoquer un trouble du drainage du liquide céphalo-rachidien.
Quelles sont les causes d'un médulloblastome ?
Un médulloblastome est principalement dû à une dégénérescence spontanée et imprévisible des cellules du système nerveux. La cause de cette croissance cellulaire soudaine et incontrôlée n'est pas encore entièrement élucidée. Cependant, les facteurs génétiques jouent un rôle identifiable. Moins de 5 % des cas sont associés à des maladies héréditaires (appelées prédispositions au cancer). Il s'agit notamment de :
- Le syndrome de Gorlin-Goltz
- Le syndrome de Rubinstein-Taybi
- Le syndrome de Turcot
- Le syndrome de Li-Fraumeni
Qui est atteint de médulloblastome ?
L'âge d'apparition du médulloblastome varie en fonction du sous-type de la tumeur. Elles apparaissent typiquement dans deux groupes d'âge : chez les jeunes enfants entre 3 et 4 ans et chez les enfants plus âgés entre 8 et 10 ans. Chez les adultes, les tumeurs sont très rares. 10 à 50 % des médulloblastomes se sont déjà propagés au moment du diagnostic. Les plus courantes sont les métastases dites «goutte à goutte». Celles-ci se propagent le long de l'espace sous-arachnoïdien ou de l'axe rachidien. Les métastases en dehors du système nerveux sont beaucoup plus rares. Ils surviennent chez environ 5 % des patients.
Quels sont les symptômes d'un médulloblastome ?
Les symptômes se développent généralement en peu de temps en raison de leur croissance rapide. Lorsque le cervelet et le 4e ventricule cérébral sont touchés par le médulloblastome, on observe des vertiges accompagnés de nausées et de vomissements, des problèmes de coordination et des troubles du mouvement tels que l'instabilité de la démarche.
Cependant, il est également possible que le tronc cérébral soit touché, ce qui peut entraîner des défaillances des nerfs crâniens. Cela se manifeste par des troubles de la vue, une vision double, une paralysie faciale, etc.
Si le drainage du liquide céphalo-rachidien est obstrué, une hydrocéphalie se produit en raison de l'accumulation de liquide céphalo-rachidien et de l'augmentation correspondante de la pression intracrânienne. Les patients souffrent alors de symptômes tels que des maux de tête, des nausées, des vomissements ou une fatigue et une somnolence marquées. En outre, une confusion et des changements de personnalité peuvent également survenir. Les nourrissons ont parfois une tête plus grosse que la moyenne et qui grandit rapidement. Cette macrocéphalie est un signe d'augmentation du liquide céphalorachidien et de la pression intracrânienne. Les enfants affectés semblent très endormis ou réagissent de manière irritable.
Les métastases spinales peuvent entraîner des douleurs et des déficits sensorimoteurs en raison de l'effet d'occupation de l'espace sur la moelle épinière.
Comment le médulloblastome est-il diagnostiqué ?
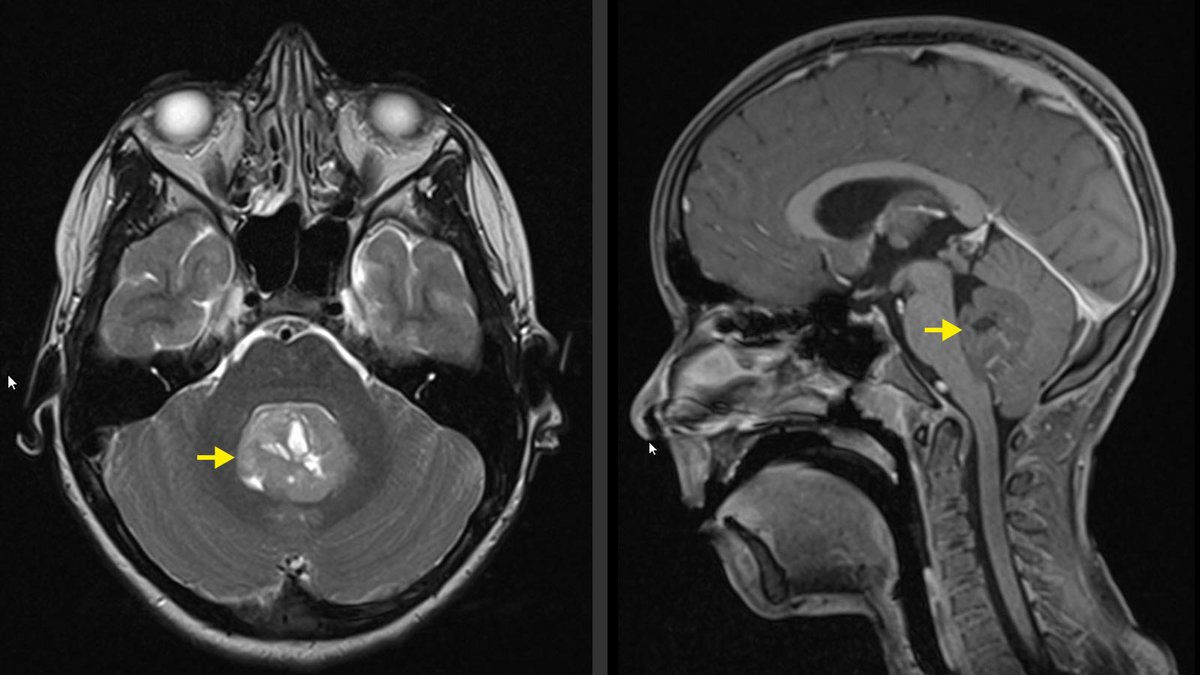
Sur une tomographie assistée par ordinateur (CT), une masse est généralement clair et réagit aux contrastes. Cependant, l'imagerie par résonance magnétique (IRM) est considérée comme la référence en matière d'imagerie et doit inclure la colonne vertébrale en plus du crâne pour exclure d'éventuelles métastases dans tout le système nerveux central.
Chez les jeunes enfants, si les fontanelles sont encore ouvertes, des indices peuvent déjà être vus à l'échographie de la tête.
En complément de l'imagerie, une ponction lombaire est effectuée pour analyser le liquide céphalorachidien, car il est parfois possible d'y déceler des cellules tumorales.
Un diagnostic exact n'est toutefois possible qu'après l'évaluation histologique et génétique moléculaire du tissu tumoral prélevé par voie chirurgicale. Des techniques avancées telles que le profilage ADN-méthylome et le séquençage de nouvelle génération (NGS) ont considérablement amélioré le diagnostic et la classification des médulloblastomes en permettant de caractériser les tumeurs au niveau *, * :
Classification
Selon la nouvelle classification de l'OMS, les médulloblastomes sont divisés en 4 sous-groupes moléculaires * , sur la base de caractéristiques génétiques et moléculaires. Cette distinction a des conséquences sur le pronostic et le traitement *:
- Activé par WNT : ces tumeurs sont associées à un très bon pronostic et présentent peu de modifications génétiques. Elles sont caractérisées par la présence de mutations dans le gène de la bêta-caténine et la monosomie 6.
- Activées par la SHH : ce groupe comprend les tumeurs caractérisées par l'activation de la voie de signalisation Sonic Hedgehog. Les tumeurs SHH associées à des mutations TP53 ont un moins bon pronostic.
- Groupe 3 et groupe 4 : ces groupes sont moins bien caractérisés que WNT et SHH, mais présentent des comportements biologiques et cliniques différents, ce qui influence le traitement et le pronostic.
En outre, d'autres sous-types doivent être différenciés : quatre sous-types de SHH- et huit sous-types de groupe 3 et de groupe 4.
Options thérapeutiques
Le traitement de choix est la résection microchirurgicale la plus complète possible de la tumeur. L'étendue de la résection est ici décisive et une ablation complète du tissu tumoral est associée à un meilleur pronostic. La planification de l'opération et du traitement qui s'ensuit est le fruit d'une collaboration intensive entre une équipe interdisciplinaire composée de neurochirurgiens, de neuroradiologues, de neuropathologues, d'oncologues et de radio-oncologues.
Le traitement postopératoire par radiothérapie crânio-spinale et/ou chimiothérapie dépend de critères individuels tels que le sous-type de la tumeur, l'âge du patient, l'état de santé général du patient, la présence de métastases et, surtout, la tolérance du patient aux mesures thérapeutiques. Chez les enfants de moins de 3 ans, une chimiothérapie intensive est souvent utilisée pour retarder la nécessité d'une radiothérapie, car celle-ci est hautement toxique et peut avoir des effets secondaires à long terme.
La collaboration entre les neurochirurgiens pédiatriques, les radio-oncologues et les oncologues pédiatriques est essentielle pour garantir un traitement optimal.
Les nouvelles découvertes concernant les sous-groupes et les sous-types pourraient entraîner à l'avenir des modifications du traitement, comme par exemple une thérapie adaptée à chaque individu avec des protocoles de traitement différents. Chez les enfants en particulier, cela pourrait permettre de minimiser les effets secondaires à long terme liés au traitement. *,*, *
Perspectives de réussite
Les chances de succès varient en fonction du sous-type de tumeur. La radiothérapie a des effets secondaires graves (déficits neuropsychologiques, endocrinopathies), ainsi qu`elle est de plus en plus substitué par la Protonthérapie, surtout chez les enfants les plus petits. L'objectif principal du traitement chirurgical est l'exérèse sûre et peu invasive de la tumeur, mais la plus radicale possible, avec rétablissement de l'écoulement libre du LCR. Si l'écoulement du LCR du patient ne peut être rétabli, une dérivation ventriculo-péritonéale dérivation ventriculo-péritonéale peut être nécessaire. Dans ce cas, le LCR est drainé dans la cavité abdominale par un cathéter placé sous la peau.
Pourquoi se faire soigner à l'Inselspital ?
Selon la localisation du médulloblastome, il existe un certain risque d'endommager les structures cérébrales voisines pendant l'opération. Pour réduire au maximum ce risque de complications, nous utilisons les procédures techniques les plus récentes telles que la neuronavigation et le neuromonitoring peropératoire. Ils permettent à nos neurochirurgiens de travailler avec un maximum de précision et de sécurité tout en réalisant une résection maximale de la tumeur. En plus, un IRM peropératoire nous permet de contrôler la radicalité de l`exérèse pendant l`opération et d`agir en cas de résidu tumoral.
-
Juraschka K, Taylor MD. Medulloblastoma in the age of molecular subgroups: a review. J Neurosurg Pediatr. 2019 Oct 1;24(4):353-363. doi: 10.3171/2019.5.PEDS18381.
-
Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, Eberhart CG, Parsons DW, Rutkowski S, Gajjar A, Ellison DW, Lichter P, Gilbertson RJ, Pomeroy SL, Kool M, Pfister SM. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012 Apr;123(4):465-72. doi: 10.1007/s00401-011-0922-z.
-
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM, Reifenberger G, Soffietti R, von Deimling A, Ellison DW. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021 Aug 2;23(8):1231-1251. doi: 10.1093/neuonc/noab106.
-
Ramaswamy V, Remke M, Bouffet E, Bailey S, Clifford SC, Doz F, Kool M, Dufour C, Vassal G, Milde T, Witt O, von Hoff K, Pietsch T, Northcott PA, Gajjar A, Robinson GW, Padovani L, André N, Massimino M, Pizer B, Packer R, Rutkowski S, Pfister SM, Taylor MD, Pomeroy SL. Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathol. 2016 Jun;131(6):821-31. doi: 10.1007/s00401-016-1569-6.
-
Maier H, Dalianis T, Kostopoulou ON. New Approaches in Targeted Therapy for Medulloblastoma in Children. Anticancer Res. 2021 Apr;41(4):1715-1726. doi: 10.21873/anticanres.14936.
-
Lutz K, Jünger ST, Messing-Jünger M. Essential Management of Pediatric Brain Tumors. Children (Basel). 2022 Apr 2;9(4):498. doi: 10.3390/children9040498.
Littérature complémentaire
- Robinson GW, Rudneva VA, Buchhalter I et al. Risk-adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol. 2018;19:768-784.
- Harbaugh R, Shaffrey CI, Couldwell WT. Neurosurgery Knowledge Update. Thieme; 2015:984.
- Greenberg MS. Handbook of Neurosurgery. Thieme; 2016:1664.
- The Neurosurgical Atlas: Medulloblastoma (free sign up).