Un hémangioblastome est une tumeur du système nerveux central, rare, bénigne et très vascularisée. Outre un nodule solide, les hémangioblastomes présentent souvent des parties kystiques. Le plus souvent, les hémangioblastomes sont localisés dans le cervelet, le tronc cérébral ou la moelle épinière. L'ablation microchirurgicale est le traitement de choix. La difficulté de l'opération varie en fonction de la taille, de la localisation et du kyste tumoral. Chez les patients présentant des hémangioblastomes multiples ou chez les patients atteints de la maladie de Von Hippel-Lindau, seuls les hémangioblastomes symptômatiques ou qui augmentent de taille sont réséqués chirurgicalement.
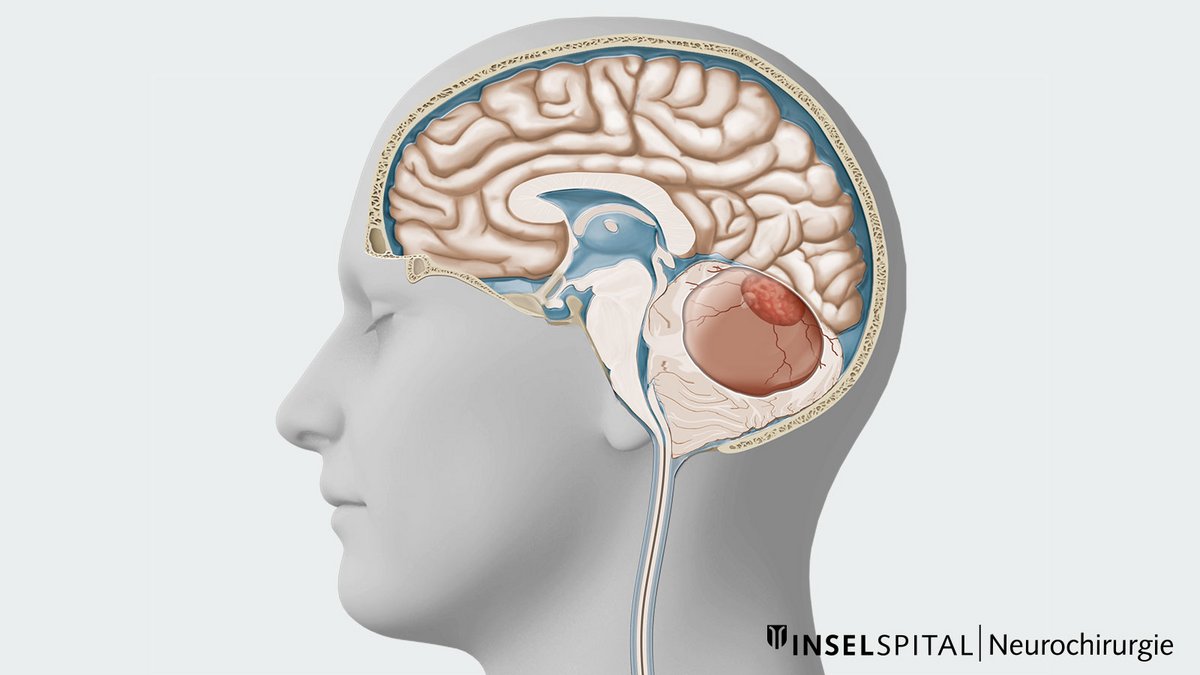
Qui est concerné par le diagnostic d’hémangioblastome ?
Les hémangioblastomes sont des tumeurs rares du système nerveux central. Les hémangioblastomes sont souvent sporadiques, mais peuvent également être liés à le syndrome de Von Hippel-Lindau (VHLS). Un bilan complémentaire est donc recommandé pour tout hémangioblastome nouvellement diagnostiqué. *
Ils représentent environ 2 % de toutes les tumeurs cérébrales primaires. 85 % sont localisés au niveau du cervelet. * Dans de rares cas, les hémangioblastomes se développent dans
- la moelle épinière cervicale (< 10 %)
- le tronc cérébral (< 5 %) ou
- le cerveau (< 2 %)
80 % des hémangioblastomes sont sporadiques et apparaissent typiquement au cours de la 4e décennie de la vie *. Les hémangioblastomes associés au VHLS se manifestent plus précocement. La plupart des patients développent la maladie entre 20 et 40 ans. *, *
Alors que les hémangioblastomes sporadiques sont généralement des tumeurs uniques, ils sont souvent multiples dans le syndrome de Von Hippel-Lindau (VHLS). Jusqu'à 84% des patients atteints de VHLS développent un hémangioblastome. *
Syndrome de Von Hippel-Lindau (VHLS)
Le syndrome de Von Hippel-Lindau a été décrit pour la première fois au début du XXe siècle par l'ophtalmologiste allemand Eugen von Hippel et le pathologue suédois Arvid Lindau. La maladie est transmise sur un mode autosomale dominante et repose sur une mutation du gène suppresseur de tumeur VHL. Cette mutation entraîne l'apparition d'une variété de tumeurs bénignes et malignes :
- hémangioblastomes du système nerveux central
- hémangiomes rétiniens
- carcinomes rénaux à cellules claires et kystes rénaux
- phéochromocytomes
- cystadénomes séreux
- tumeurs neuroendocrines du pancréas
- tumeurs endolymphoïdes de l'oreille moyenne
- cystadénomes papillaires du testicule
Pour un diagnostic de VHLS, une ou plusieurs tumeurs doivent être présentes, en fonction des antécédents familiaux. Des tests génétiques sont également prévus. En général, une évaluation génétique impliquant toute la famille est d'une grande importance dans cette maladie héréditaire. *
Quels sont les symptômes d'un hémangioblastome ?
Lorsqu'un hémangioblastome se développe et comprime localement le parenchyme cérébral, cela peut porter à des symptômes neurologiques. Les hémorragies aiguës sont rares. Les hémangioblastomes provoquent plutôt des symptômes typiques d'une atteinte du cervelet. Les patients signalent en principe des symptômes tels que maux de tête, nausées, vomissements, troubles de la démarche et de l'équilibre, vision double et troubles de la motricité fine. Ces déficits sont dus à des lésions directes du tissu cérébelleux, mais aussi en partie à la pression exercée sur le tronc cérébral ou à l'augmentation de la pression intracrânienne causée par une obstruction de l’écoulement du liquide céphalo-rachidien. *
Les hémangioblastomes de la moelle épinière ou du tronc cérébra, moins fréquents, peuvent être responsables de déficits neurologiques similaires. Les hémangioblastomes dans la région de la moelle épinière peuvent en outre provoquer des troubles sensoriels et moteurs au niveau des extrémités.
Comment diagnostique-t-on un hémangioblastome ?
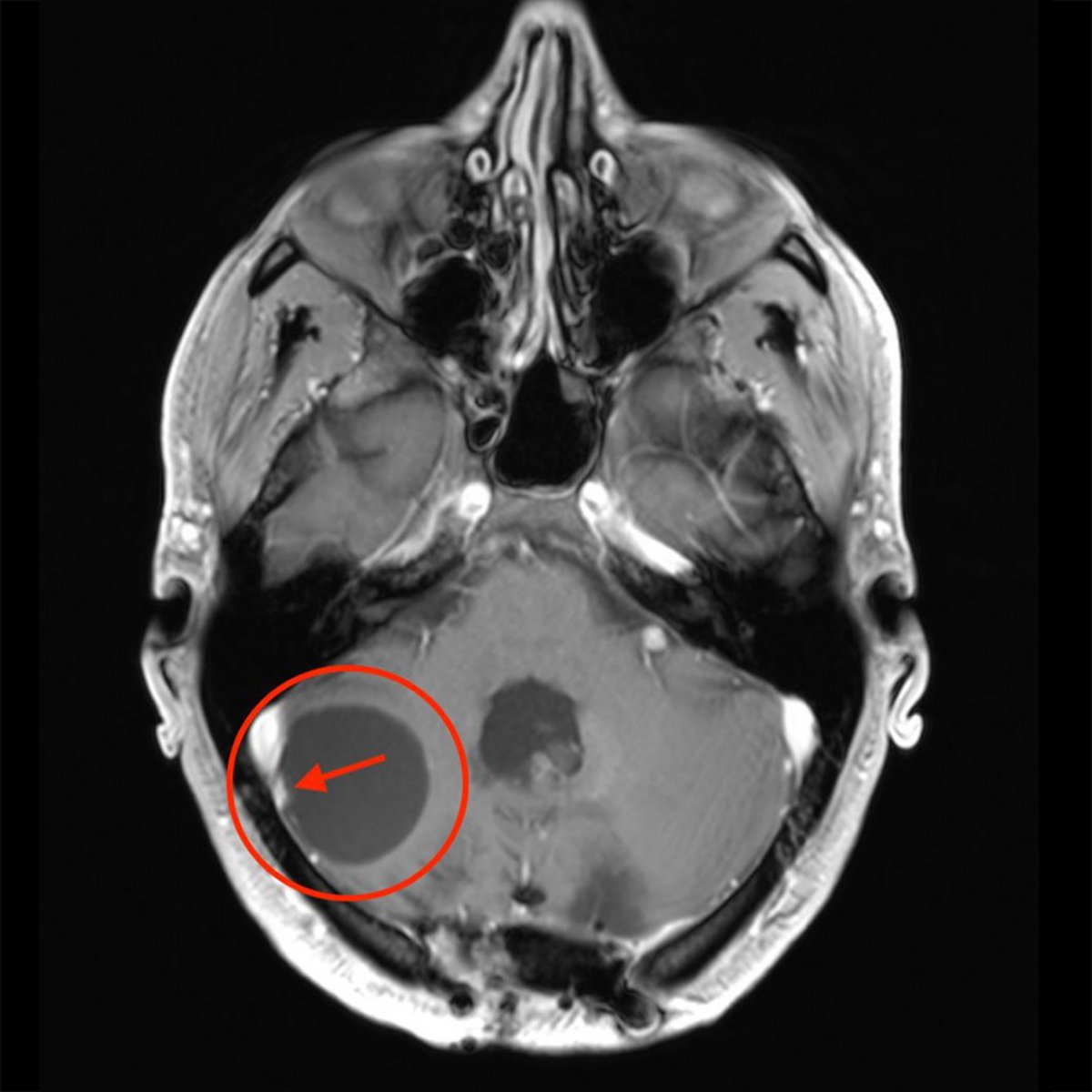
En cas de symptômes neurologiques, un examen radiologique est effectué au moyen d'une tomographie computérisée (CT) ou d'une imagerie par résonance magnétique (IRM). L'IRM est la meilleure méthode en raison de sa résolution plus élevée et de l'imagerie plus détaillée au niveau cérébral. L'imagerie révèle souvent une masse kystique et, après administration de produit de contraste, un petit nodule tumoral solide.
En plus du CT scanner ou de l'IRM, une angiographie par soustraction numérique (DSA) peut également être réalisée. Dans ce cas, un fin cathéter est inséré dans une artère de l'aine et est acheminé jusqu'au cerveau. Là, les vaisseaux cérébraux sont ensuite imagés par des injections de contraste. La DSA peut être nécessaire pour détecter les plus petites parties solides de la tumeur qui ne peuvent pas être visualisées sur le CT ou l'IRM, ou pour visualiser la vascularisation de l'hémangioblastome. Cependant, le diagnostic définitif d'hémangioblastome nécessite un examen histologique des échantillons de tissus prélevés au moment de la chirurgie.
Quelles sont les options de traitement?
Microchirurgie
L'ablation microchirurgicale douce de l'hémangioblastome est le traitement de choix. Au cours de l'opération, le nodule de la tumeur solide est retiré et le kyste est drainé. L'ablation de la paroi du kyste n'est pas nécessaire. Dans le cas d'hémangioblastomes individuels, la chirurgie est une thérapie curative, c'est-à-dire qu'aucune autre thérapie telle que la radiothérapie ou la chimiothérapie n'est nécessaire après la chirurgie. 82 à 98 % des patients présentent une amélioration après l'ablation chirurgicale d’un hémangioblastome.
Les hémangioblastomes associés au VHLS récidivent plus fréquemment et sont souvent multiples. Pour cette raison, ils ne sont retirés chirurgicalement que si symptomatiques, s'ils se développent rapidement ou si leur croissance menace des structures cérébrales importantes. *
Radiochirurgie
La radiochirurgie stéréotaxique ou l'irradiation fractionnée sont utilisées pour les tumeurs inopérables ou les tumeurs multiples situées à plusieurs endroits différents afin d'éviter plusieurs interventions chirurgicales. *
-
Kuharic M, Jankovic D, Splavski B, Boop F, Arnautovic K. Hemangioblastomas of the Posterior Cranial Fossa in Adults: Demographics, Clinical, Morphologic, Pathologic, Surgical Features, and Outcomes. A Systematic Review. World Neurosurgery. 2018;110:e1049-e1062.
-
Lonser R, Glenn G, Walther M, Chew E, Libutti S, Linehan W et al. von Hippel-Lindau disease. The Lancet. 2003;361(9374):2059-2067.
-
Bamps S, Calenbergh F, Vleeschouwer S, Loon J, Sciot R, Legius E et al. What the neurosurgeon should know about hemangioblastoma, both sporadic and in Von Hippel-Lindau disease: A literature review. Surgical Neurology International. 2013;4(1):145.
-
Wanebo J, Lonser R, Glenn G, Oldfield E. The natural history of hemangioblastomas of the central nervous system in patients with von Hippel—Lindau disease. Journal of Neurosurgery. 2003;98(1):82-94.
-
Bridges K, Jaboin J, Kubicky C, Than K. Stereotactic radiosurgery versus surgical resection for spinal hemangioblastoma: A systematic review. Clinical Neurology and Neurosurgery. 2017;154:59-66.
-
Vaganovs P, Bokums K, Miklaševics E, Plonis J, Zarina L, Geldners I et al. Von Hippel-Lindau Syndrome: Diagnosis and Management of Hemangioblastoma and Pheochromocytoma. Case Reports in Urology. 2013;2013:1-5.