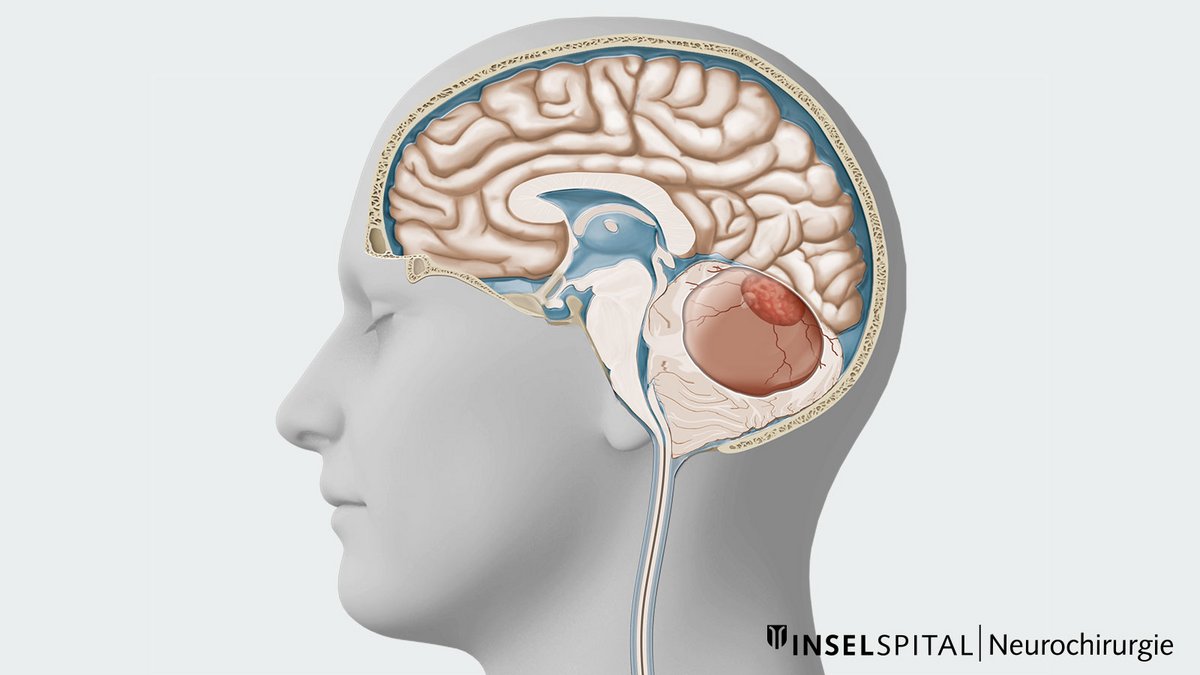
Ein Hämangioblastom ist ein seltener, gutartiger und äusserst gefässreicher Tumor des zentralen Nervensystems. Neben einem soliden Knoten weisen Hämangioblastome häufig auch zystische Anteile auf. Meist findet man Hämangioblastome im Kleinhirn, Hirnstamm oder Rückenmark. Die mikrochirurgische Entfernung ist die Therapie der ersten Wahl. Die Schwierigkeit dieser Operation hängt dabei von der Grösse, Lage und Beschaffenheit des Tumors ab. Bei Patienten mit mehreren Hämangioblastomen oder bei Patienten mit der Von-Hippel-Lindau-Erkrankung operiert man nur die Tumoren, die Beschwerden verursachen oder wachsen.
Wer erkrankt an einem Hämangioblastom?
Hämangioblastome sind seltene Tumoren des zentralen Nervensystems. Sie treten häufig sporadisch auf, können aber auch auf das Von-Hippel-Lindau-Syndrom (VHLS) zurückzuführen sein. Aus diesem Grund sollten bei jedem neu diagnostizierten Hämangioblastom weitere Abklärungen durchgeführt werden. *
Hämangioblastome machen etwa 2 % aller primären Hirntumoren aus. Zu 85 % sind sie im Kleinhirn lokalisiert. * Selten wachsen Hämangioblastome auch an anderen Orten:
- im Bereich des zervikalen Rückenmarks (< 10 %)
- des Hirnstamms (< 5 %)
- des Grosshirns (< 2 %)
80 % aller Hämangioblastome sind sporadisch und treten typischerweise in der 4. Lebensdekade auf. * Die Manifestation von VHLS-assoziierten Hämangioblastomen ist früher. Die meisten Patienten erkranken zwischen dem 20. und 40. Lebensjahr. *, *
Während es sich bei sporadischen Hämangioblastomen meistens um einzelne Tumoren handelt, treten sie im Rahmen des Von-Hippel-Lindau-Syndroms (VHLS) häufig multipel auf. Bis zu 84 % von Patienten mit einem VHLS erkranken an einem Hämangioblastom. *
Von-Hippel-Lindau-Syndrom (VHLS)
Das Von-Hippel-Lindau-Syndrom wurde erstmalig zu Beginn des 20. Jahrhunderts von dem deutschen Augenarzt Eugen von Hippel und dem schwedischen Pathologen Arvid Lindau beschrieben. Die Erkrankung wird autosomal dominant vererbt und beruht auf einer Mutation des Tumorsuppressor-Gens VHL. Die Mutation führt zum Auftreten einer Vielzahl von gutartigen und bösartigen Tumoren:
- Hämangioblastome des zentralen Nervensystems
- Hämangiome der Retina
- klarzellige Nierenzellkarzinome und Nierenzysten
- Phäochromozytome
- seröse Zystadenome
- neuroendokrine Tumoren des Pankreas
- endolymphatische Tumoren des Mittelohrs
- papilläre Zystadenome des Hodens
Für die Diagnose eines VHLS müssen je nach Familienanamnese ein oder mehrere Tumoren vorliegen. Es stehen auch genetische Tests zur Verfügung. Generell ist bei dieser Erbkrankheit eine genetische Beratung unter Einbezug der gesamten Familie von grosser Wichtigkeit. *
Welche Symptome verursacht ein Hämangioblastom?
Wenn ein Hämangioblastom wächst und lokal das Hirngewebe komprimiert, kann es Beschwerden hervorrufen. Akute Blutungen sind eher selten. Hämangioblastome verursachen vielmehr Symptome, die typisch sind für eine Beeinträchtigung des Kleinhirns. Patienten berichten über Beschwerden wie Kopfschmerzen, Übelkeit, Erbrechen, Gang- und Gleichgewichtsstörungen, Doppelbilder und Störungen der Feinmotorik. Diese Ausfälle sind zum Teil auf eine direkte Schädigung des Kleinhirngewebes zurückzuführen, teils auch bedingt durch Druck auf den Hirnstamm oder durch einen insgesamt erhöhten Schädeldruck durch eine Stauung des Hirnwassers. *
Seltenere Hämangioblastome im Bereich des Rückenmarks oder des Hirnstamms verursachen ähnliche neurologische Ausfälle. Hämangioblastome im Bereich des Rückenmarks können zusätzlich zu Gefühlsstörungen in den Extremitäten führen.
Wie wird ein Hämangioblastom diagnostiziert?
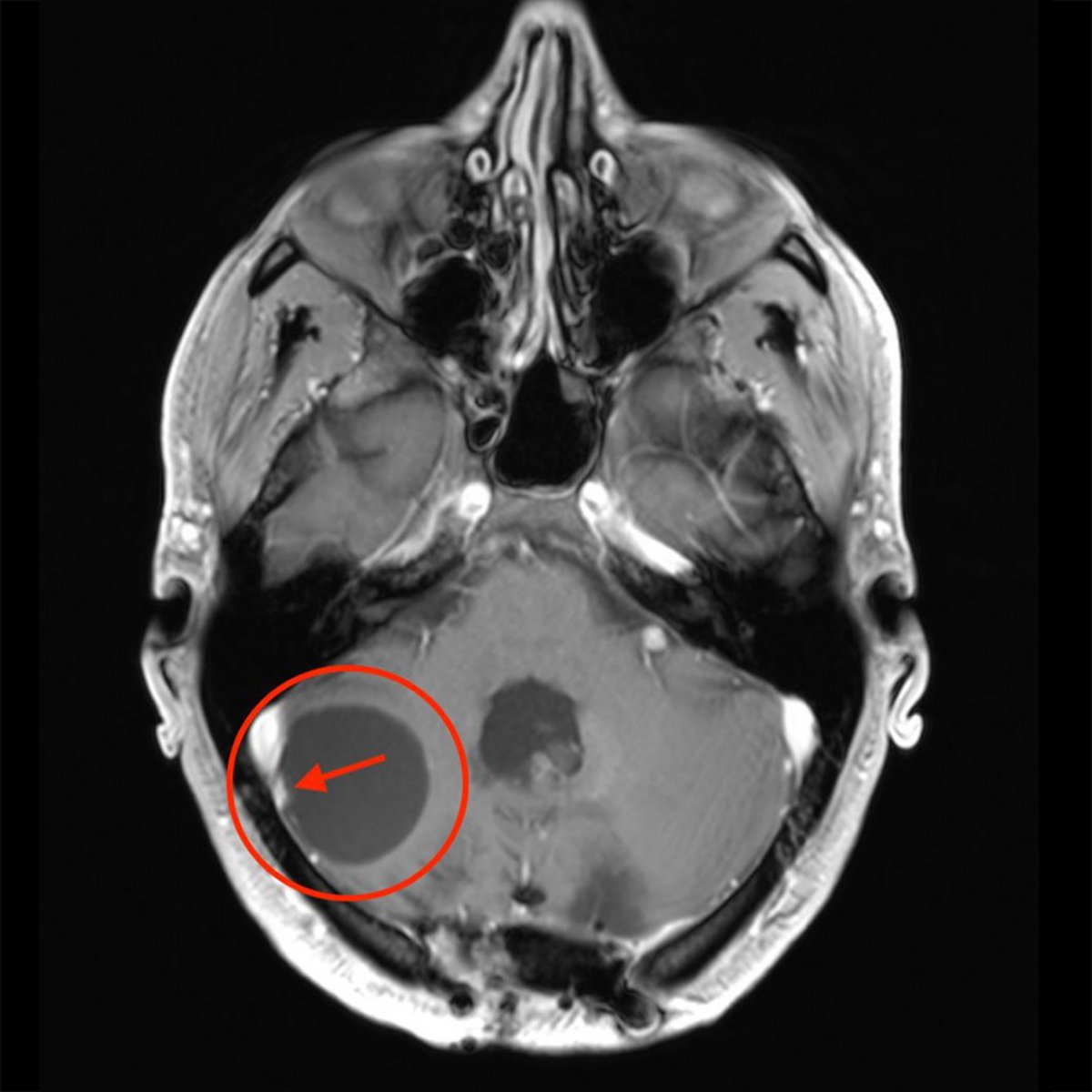
Bei entsprechenden neurologischen Beschwerden wird eine radiologische Abklärung mittels Computertomografie (CT) oder Magnetresonanztomografie (MRT oder MRI von engl. Magnetic Resonance Imaging) vorgenommen. Das MRI ist aufgrund der höheren Auflösung und detaillierteren Weichteildarstellung dabei die bessere Methode. In der Bildgebung findet sich häufig eine zystische Raumforderung und nach Gabe von Kontrastmittel nur ein kleiner solider Tumorknoten. Die Grösse des soliden und zystischen Anteils kann bei Hämangioblastomen jedoch stark variieren.
Neben CT oder MRI kann zusätzlich auch eine digitale Subtraktionsangiografie (DSA) durchgeführt werden. Hierbei wird ein dünner Katheter in die Arterie in der Leiste eingeführt und bis zum Gehirn vorgeschoben. Dort werden dann die zerebralen Gefässe über Kontrastmittelinjektionen dargestellt. Eine DSA kann notwendig sein, um kleinste solide Tumoranteile nachzuweisen, die im CT oder MRI nicht darstellbar sind, oder um die Vaskularisierung des Hämangioblastoms darzustellen. Für die definitive Diagnose eines Hämangioblastoms braucht es jedoch die feingewebliche Untersuchung der Gewebeproben nach der Operation.
Welche Behandlungsmöglichkeiten gibt es?
Mikrochirurgie
Die schonende mikrochirurgische Entfernung des Hämangioblastoms ist die Therapie der Wahl. Bei der Operation werden, der solide Tumorknoten entfernt und die Zyste entlastet. Ein Entfernen der Zystenwand ist nicht notwendig. Bei einzelnen Hämangioblastomen ist die Operation eine kurative Therapie, d. h. nach der Operation sind keine weiteren Therapien wie Bestrahlung oder Chemotherapie notwendig. 82–98 % der Patienten zeigen nach der operativen Entfernung des Hämangioblastoms eine Besserung der Beschwerden.
VHLS-assoziierte Hämangioblastome rezidivieren häufiger und treten oft multipel auf. Aus diesem Grund werden sie nur dann chirurgisch entfernt, wenn sie Beschwerden verursachen, stark wachsen oder wichtige Hirnstrukturen durch ihr Wachstum bedrohen. *
Radiotherapie
Eine stereotaktische Radiochirurgie oder fraktionierte Bestrahlung werden bei nicht operablen Tumoren oder bei multiplen Tumoren in verschiedenen Lokalisationen eingesetzt, um mehrfache operative Eingriffe zu vermeiden. *
Referenzen
-
Kuharic M, Jankovic D, Splavski B, Boop F, Arnautovic K. Hemangioblastomas of the Posterior Cranial Fossa in Adults: Demographics, Clinical, Morphologic, Pathologic, Surgical Features, and Outcomes. A Systematic Review. World Neurosurgery. 2018;110:e1049-e1062.
-
Lonser R, Glenn G, Walther M, Chew E, Libutti S, Linehan W et al. von Hippel-Lindau disease. The Lancet. 2003;361(9374):2059-2067.
-
Bamps S, Calenbergh F, Vleeschouwer S, Loon J, Sciot R, Legius E et al. What the neurosurgeon should know about hemangioblastoma, both sporadic and in Von Hippel-Lindau disease: A literature review. Surgical Neurology International. 2013;4(1):145.
-
Wanebo J, Lonser R, Glenn G, Oldfield E. The natural history of hemangioblastomas of the central nervous system in patients with von Hippel—Lindau disease. Journal of Neurosurgery. 2003;98(1):82-94.
-
Bridges K, Jaboin J, Kubicky C, Than K. Stereotactic radiosurgery versus surgical resection for spinal hemangioblastoma: A systematic review. Clinical Neurology and Neurosurgery. 2017;154:59-66.
-
Vaganovs P, Bokums K, Miklaševics E, Plonis J, Zarina L, Geldners I et al. Von Hippel-Lindau Syndrome: Diagnosis and Management of Hemangioblastoma and Pheochromocytoma. Case Reports in Urology. 2013;2013:1-5.