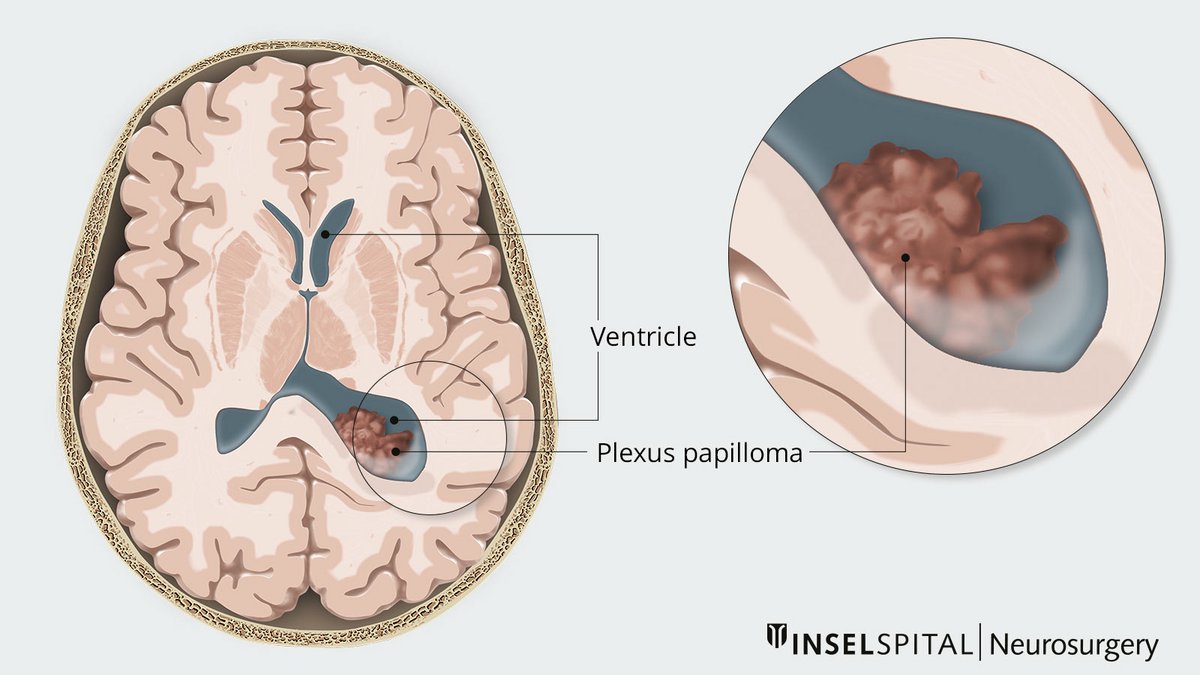
Plexus tumors are very rare brain tumors that develop from the choroid plexus, a network of epithelial cells that line the ventricular system and produce cerebrospinal fluid (CSF). The cells of plexus tumors can grow with varying degrees of aggressiveness. The majority of benign tumors are plexus papillomas, while malignant tumors are plexus carcinomas. The tumor occurs mainly in children and adolescents – more than 70% of plexus papillomas develop before the age of 3. The treatment of choice is complete resection, which may be followed by other necessary therapeutic steps.
What types of plexus tumors are there?
There are benign as well as malignant plexus tumors. According to the WHO classification, there are basically 3 degrees of severity for these tumors:
- benign choroid plexus papilloma (CPP): WHO grade I
- intermediate grade malignant atypical choroid plexus papilloma (APP): WHO grade II
- higher grade malignant choroid plexus carcinoma (CPC): WHO grade III
The different grades are distinguished on the basis of their fine-tissue characteristics and their mitotic activity, i.e. their cell division processes and cell reproduction. Imaging also reveals typical appearances of the individual tumors.
All plexus tumors can scatter in the body via what are known as drop metastases.
There is a difference in tumor location depending on the age of onset. In children, a supratentorial location in the left ventricle is usually observed. In adults, however, the tumor usually grows in the 4th ventricle and is thus localized infratentorially. However, ectopic occurrence in the brain parenchyma, suprasellar and spinal has also been described.
How common is a plexus tumor?
Plexus papillomas account for less than 0.5% of intracranial tumors. Thus, they are quite rare compared to other brain tumors such as gliomas. The annual incidence is estimated at approximately 0.3 per 1 000 000 population. They are slightly more common in males than in females, somewhat at a ratio of 1.2 : 1.
Plexus carcinomas are diagnosed even less frequently compared to plexus papillomas (at a ratio of 1 : 5). Plexus tumors occur primarily in infancy and childhood. The average age at diagnosis is 3.5 years.
What are the causes of a plexus tumor?
Plexus tumor usually develops sporadically. However, patients with Li-Fraumeni syndrome are known to develop choroid plexus carcinoma more frequently. The cause is thought to be a TP53 mutation. This mutation of the tumor suppressor protein TP53 can also occur in other genetic diseases or sporadically.
Apart from hereditary factors, alterations of certain genes or chromosomes seem to play an important role in the development of plexus tumors. Modern genetic analyses have identified 6 corresponding genes (TWIST1, WIF1, AJAP1, BCLAF1, TRPM3, IL6ST).
Also, a connection with a specific signal transduction pathway is discussed, which is involved in the development of gliomas and embryonic tumors. This is known as the Notch pathway.
Furthermore, the methylation of certain gene segments is being investigated, which is associated with the loss of a tumor suppression function.
What are the symptoms of a plexus tumor?
There is no typical symptomatology by which a plexus tumor can be reliably diagnosed. However, depending on the localization of the tumor, typical signs occur:
- Hydrocephalus
Since plexus tumors arise from the choroid plexus, which is also responsible for the formation of cerebrospinal fluid (CSF), the tumor cells of these tumors also form increased amounts of CSF. Due to the localization of the plexus tumor, normal CSF flow is often additionally restricted. A major symptom is therefore hydrocephalus due to a buildup of cerebrospinal fluid. Such hydrocephalus is present in up to 80% of patients. - Cranial Pressure Symptomatology
The accumulated cerebrospinal fluid increases intracranial pressure, causing very typical symptoms. The signs of intracranial pressure include severe headache, nausea and vomiting. In addition, papilledema, an edema in the tissue of the optic nerve papilla, may occur. Another severe symptom of increased intracranial pressure is impaired consciousness. - Additional Signs
Depending on the location of the tumor, other symptoms such as cranial nerve loss or paralysis may occur. - Macrocephaly
In children, another symptom is macrocephaly. This refers to an above-average size of the skull compared to the rest of the body. This is caused by a separation of the cranial sutures (sutures) and tense fontanelles in the child's head.
How is a plexus tumor diagnosed?
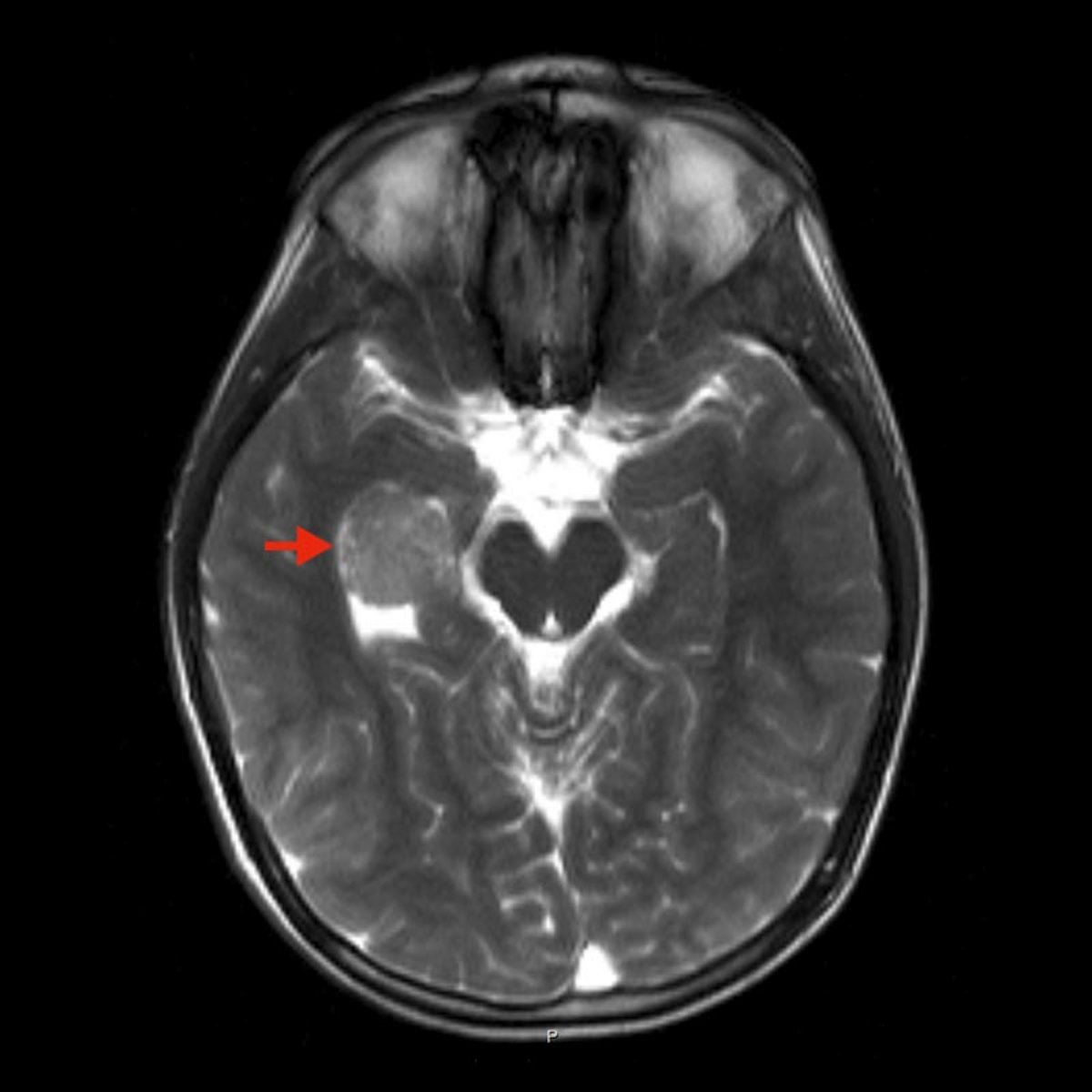
Diagnosis is initially based on imaging procedures. Magnetic resonance imaging (MRI) of the skull is the gold standard. Here, after contrast medium administration, a typical cauliflower-like contrast medium-absorbing structure is seen in the area of the ventricular system. Multiple flow voids can also be observed in additional sequences.
In addition, calcifications can very often be visualized in computed tomography (CT).
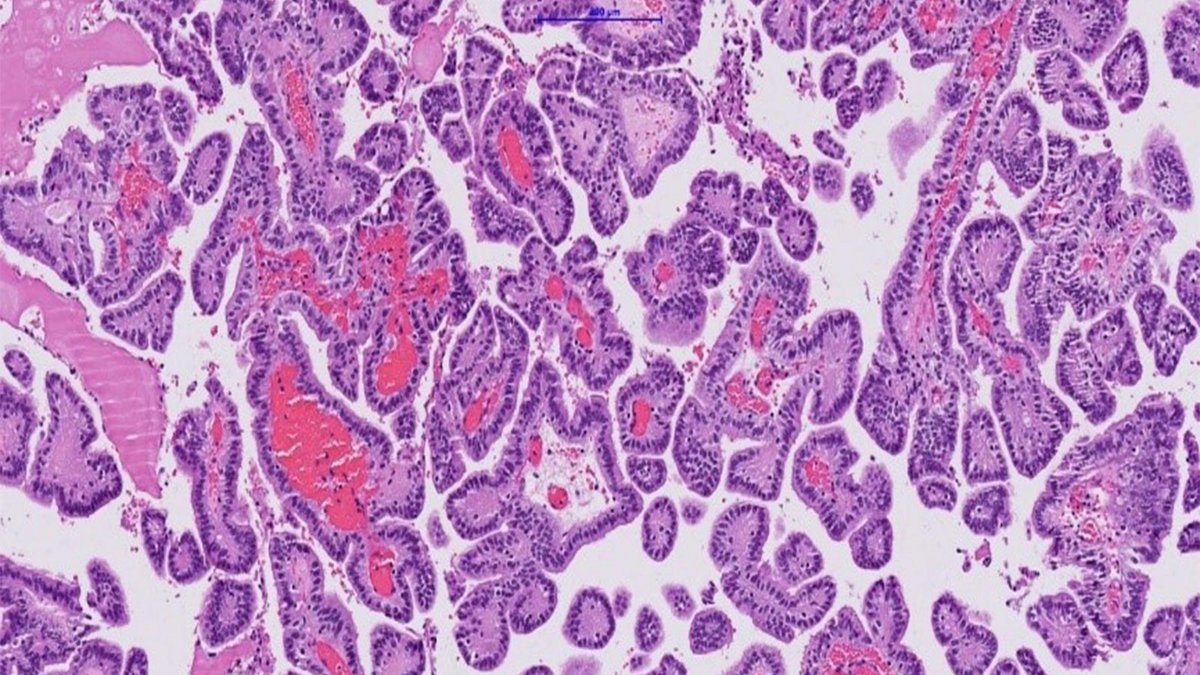
The definitive diagnosis can ultimately only be made via a histopathological examination of the tumor tissue, so that a tissue collection is always necessary.
How is a plexus tumor treated?
Complete removal of the tumor is always the main goal. This is performed at Inselspital using gentle microsurgical or endoscopic procedures.
Supporting techniques are used for planning and performing the operation, which are designed to further improve accuracy, tissue protection and safety. These include neuronavigation, intraoperative monitoring, and intraoperative imaging techniques.
Due to the mostly intraventricular location of the tumors, different access routes are possible with different chances and risks, so that the respective surgical procedure has to be planned individually for each patient
In case of existing hydrocephalus due to CSF congestion, the cerebrospinal fluid outflow must be restored. In this case, preoperative occlusion of blood vessels (embolization) may also be considered.
In case of WHO grade I tumor, complete resection of the plexus papilloma means cure for the patient. Therefore, in case of recurrent tumor or residual tumor, repeated surgery is recommended. Prognosis is very good after complete resection of plexus papilloma and recurrence of tumor is very rare. Meta-analyses show 1-, 5-, and 10-year survival rates of 90%, 81%, and 77%, respectively. The high survival in the new studies is due to modern surgical procedures that allow more complete resection.
For WHO grade II and III plexus tumors, adjunctive radio-chemotherapy should be evaluated. The 5-year recurrence rate for a WHO grade II plexus tumor is 5-fold increased compared to a WHO grade I plexus papilloma. Therefore, continuation of treatment with radiation or chemotherapy is usually necessary for these tumors. The number of studies is too small to make clear recommendations for a specific chemotherapeutic agent, so we always make an individual decision within the framework of our interdisciplinary neuro-oncology boards.
Why you should seek treatment at Inselspital
At Inselspital, the best possible treatment strategy is determined individually for each patient. This is done in our certified neuro-oncological tumor center by an interdisciplinary team. This is known as the Tumor Board, which meets weekly and is made up of specialists from neurosurgery, neurology, neuro-oncology, nuclear medicine, radio-oncology and pathology. Here, each patient is discussed individually in order to determine the optimal treatment options for him or her.
For the operation itself, we use innovative technical procedures such as neuronavigation and intraoperative neuromonitoring. These new achievements are a guarantee for maximum precision during the operation and the highest safety for our patients.
Further reading
- Safaee M, Oh MC, Bloch O, Sun MZ, Kaur G, Auguste KI, Tihan T, Parsa AT. Choroid plexus papillomas: advances in molecular biology and understanding of tumorigenesis. Neuro Oncol. 2013 Mar;15(3):255-67.
- Safaee M, Clark AJ, Bloch O, Oh MC, Singh A, Auguste KI, Gupta N, McDermott MW, Aghi MK, Berger MS, Parsa AT. Surgical outcomes in choroid plexus papillomas: an institutional experience. J Neurooncol. 2013 May;113(1):117-25.
- Wolburg H, Paulus W. Choroid plexus: biology and pathology. Acta Neuropathol.2010 Jan;119(1):75-88.