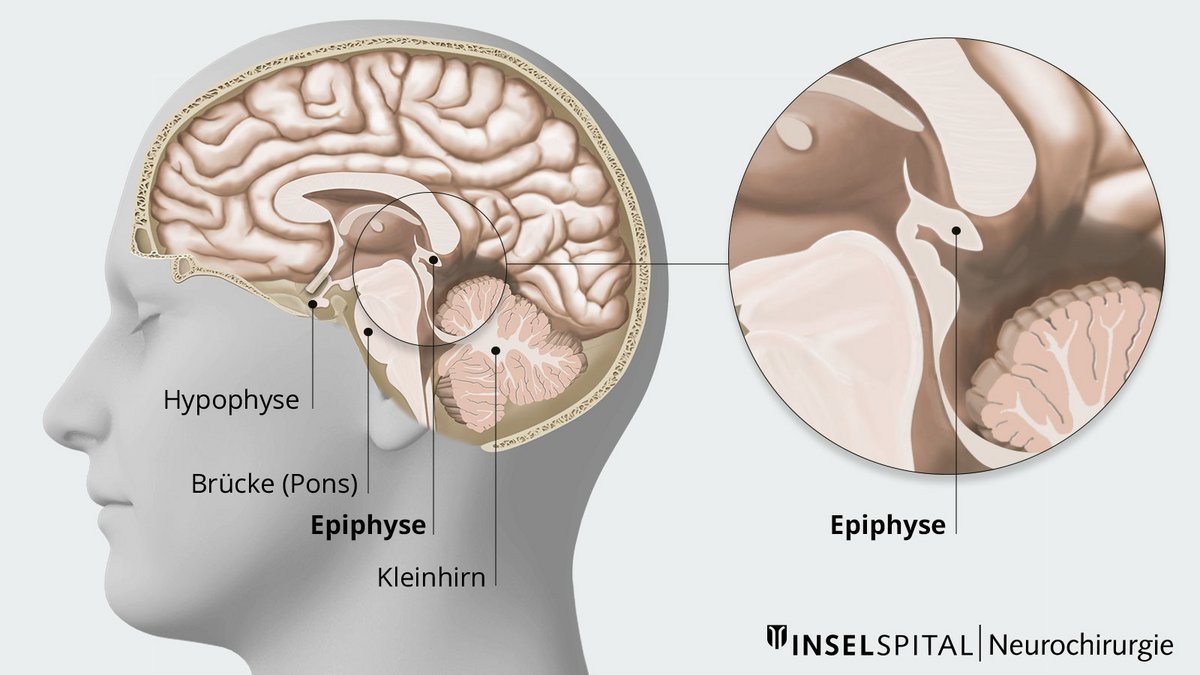
Die Tumoren der Pinealisregion bezeichnet man als Pinealistumoren, Tumoren der Zirbeldrüse oder Pinealome. Es handelt sich bei ihnen um eher seltene Tumoren. Die Zirbeldrüse oder auch Epiphyse ist eine kleine, endokrine Drüse auf der Rückseite des Mittelhirns. Sie grenzt an den 3. Ventrikel, einem Teil des Liquorabflusssystems. Die Zirbeldrüse produziert das Hormon Melatonin und ist damit hauptsächlich verantwortlich für die Steuerung des Schlaf-Wach-Rhythmus des menschlichen Körpers.
Wer ist am häufigsten von einem Pinealistumor betroffen?
Pinealistumoren machen 3–8 % der Hirntumoren bei Kindern im Alter von 1–12 Jahren aus. Bei Erwachsenen sind sie für weniger als 1 % der Hirntumoren verantwortlich, wobei Männer häufiger betroffen sind als Frauen.
Es gibt mehr als 17 verschiedene Tumorarten in der Pinealisregion. Am häufigsten findet man Germinome (50–75 %), gefolgt von Astrozytomen, Teratomen und Pinealoblastomen. Einige Tumorarten wie Germinalzelltumoren, Ependymome und Pinealzelltumoren streuen häufig über den Liquorabfluss und führen zu sogenannten Abtropfmetastasen *, *, *, *.
Welche Symptome verursacht ein Pinealistumor?
Aufgrund der anatomischen Lage der Glandula pinealis kommt es bei Raumforderungen in dieser Gegend typischerweise zu Druck auf das umgebende Hirngewebe (Masseneffekt) und damit zu Beschwerden:
- Leitsymptom ist ein Aufstau des Hirnwassers, auch Hydrozephalus genannt. Patienten leiden dann in der Regel unter Kopfschmerzen, Übelkeit, Müdigkeit oder Gedächtnisstörungen.
- Selten kann es auch zu akuten Kopfschmerzen kommen aufgrund einer plötzlichen Einblutung oder einer Durchblutungsstörung, die durch den Tumor verursacht werden.
- Kommt es zu einer Kompression des Hirnstamms können Patienten Doppelbilder oder andere Augenbewegungsstörungen bemerken.
- Mitunter kann es auch zu hormonellen Störungen kommen, wenn der Tumor Druck auf die Hirnanhangsdrüse (Hypophyse) ausübt.
- Im Fall von Abtropfmetastasen in der Wirbelsäule, können diese das Rückenmark oder Nervenwurzeln einengen und zu neurologischen Ausfällen und Schmerzen führen *, *, *.
Wie wird ein Pinealistumor diagnostiziert?
Bildgebung
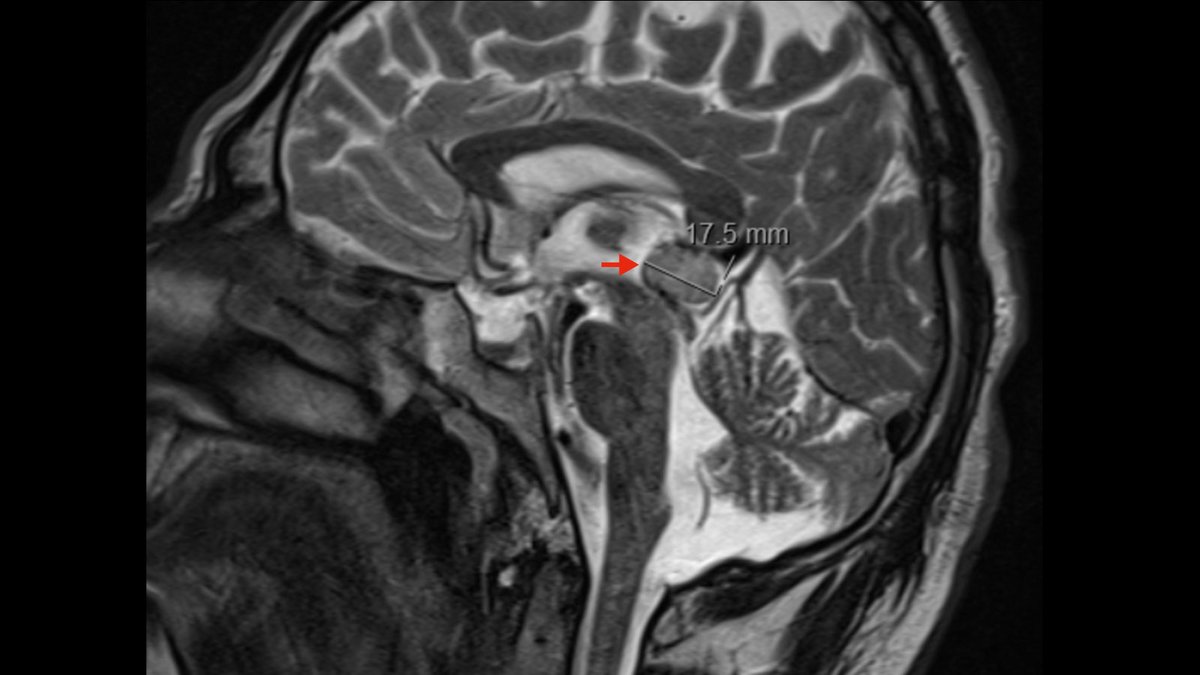
Als Goldstandard gilt die Magnetresonanztomografie (MRT bzw. MRI von engl. Magnetic Resonance Imaging), bei der sich Auffälligkeiten in der Pinealisgegend zeigen.
Mit Hilfe von Kontrastmittel kann gelegentlich die Zystenwand einer Pinealiszyste dargestellt werden. Pinealiszysten sind sehr häufige gutartige Veränderungen im Bereich der Zirbeldrüse. Irregularitäten der Zystenwand wie knotenartige Veränderungen können jedoch auf eine potenziell bösartige Raumforderung hinweisen.
Ungenügend für die Diagnosestellung ist die Computertomografie (CT), bei der das Signal der Zyste häufig dem des Liquors entspricht und deshalb auch eine falsche Diagnose liefern kann.
Zudem empfiehlt sich die Komplettierung der bildgebenden Diagnostik mit ergänzenden MRI-Aufnahmen der gesamten Wirbelsäule zum Ausschluss von Abtropfmetastasen *, *.
Tumormarker
Einige Pinealistumoren wie die Keimzelltumoren können erhöhte Tumormarker (Beta-hCG, Alpha-Fetoprotein, plazentare alkalische Phosphatase) im Liquor und Blut produzieren. In Fällen mit positiven Tumormarkern können regelmässige laborchemische Verlaufskontrollen durchgeführt werden, um den Behandlungserfolg zu beurteilen oder ein Rezidiv zu diagnostizieren. Zur Diagnosestellung reichen die Tumormarker jedoch nicht aus, da viele Tumoren der Pinealisregion aus gemischten Zellen bestehen und nicht zwingend erhöhte Tumormarker aufweisen müssen *, *, *.
Histologie
Zur definitiven Diagnosesicherung muss das Tumorgewebe histologisch analysiert werden. Zunächst wird Gewebe des Tumors operativ mittels Resektion oder Biopsie entnommen. Das Gewebe wird anschliessend von einem Pathologen aufgearbeitet. Dafür wird es mit Spezialfärbungen eingefärbt und unter dem Mikroskop analysiert und ausgewertet *, *,*, *. Bis zum Erhalt der feingeweblichen Untersuchungsergebnisse dauert es im Schnitt 3–5 Tage.
Wie werden Pinealistumoren behandelt?
Es gibt eine Vielzahl verschiedener Arten von Pinealistumoren, die eine unterschiedliche Therapie erfordern. Bei uns am Inselspital wird die konkrete Therapie von unserem interdisziplinären Tumorboard bestehend aus Neurochirurgen, Neurologen, Neuroradiologen, Onkologen und Radioonkologen festgelegt – abhängig von den Ergebnissen aus der feingeweblichen Untersuchung und den Tumormarkern.
Pinealzelltumoren
Hier gibt es zum einen das Pineozytom, einen gut differenzierten, gutartigen Tumor. Bei einem Pineozytom ist die operative Entfernung die Behandlung der Wahl. Wenn eine vollständige Resektion nicht möglich ist, folgt nach der Operation eine ergänzende Radiotherapie.
Zum anderen gibt es das Pineoblastom, einen bösartigen Tumor, der zu den primitiven neuroektodermalen Tumoren zählt (PNET).
Sowohl das Pineozytom als auch das Pineoblastom können über den Liquorabfluss metastasieren und reagieren gut auf eine Radiotherapie. Bei Kindern unter 3 Jahren wird allerdings von einer Radiotherapie abgesehen. Der Grund sind die erhöhten Nebenwirkungen auf das sich noch entwickelnde Gehirn.
Ohne Behandlung zeigen 91 % der Patienten innerhalb von 2 Jahren ein Tumorwachstum. Dagegen weisen über 50 % der behandelten Patienten nach 5 Jahren weiterhin stabile Verhältnisse auf *, *, *.
Keimzelltumoren
Keimzelltumoren werden unterschieden in Germinome und nicht-germinomatöse Keimzelltumoren (NGGCT für engl. nongerminomatous germ cell tumor).
Germinome benötigen in der Regel keine Resektion und können nur mit Radiotherapie behandelt werden. Hierbei zeigt sich eine 10-Jahres-Überlebensrate von > 90 %.
Bei den NGGCT unterscheidet man weiter zwischen Embryonalkarzinomen, Choriokarzinomen und endodermalen Sinustumoren (Dottersacktumoren). Diese werden mittels Radiochemotherapie behandelt, einer Kombination aus Strahlentherapie und Chemotherapie *, *, *.
Pinealiszysten
Bei Pinealiszysten handelt es sich um gutartige Raumforderungen, die in der Regel keine Symptome hervorrufen, solange sie nicht grösser als 1–2 cm sind. Bei etwa 4 % aller durchgeführten MRI vom Kopf findet man als Zufallsbefund eine Pinealiszyste. Bei Kindern werden aufgrund des möglichen Zystenwachstums regelmässige bildgebende Kontrollen durchgeführt. Bei Erwachsenen zeigen sich die Zysten in der Regel stabil, so dass eine Verlaufskontrolle per MRI nur bei Auftreten von Symptomen empfohlen wird. Wenn die Zysten wachsen oder Beschwerden verursachen, wird die operative Resektion empfohlen. Die Erfolgsrate der Operation liegt bei 90–95 % *, *, *.
Operationsablauf
Für die Operation wird der Kopf des Patienten mit einer 3-Punkt-Halterung fixiert. Mit Hilfe der Neuronavigation werden die aktuellsten MRI-Bilder des Patienten mit der aktuellen fixierten Kopflage abgeglichen.
Nach dem Hautschnitt wird der Schädelknochen mit einer speziellen Knochensäge eröffnet. Am häufigsten geschieht dies von hinten, oberhalb des Kleinhirns in der Grenze zwischen Gross- und Kleinhirn (infratentorieller suprazerebellärer Zugang). In der Regel kann von der Mittellinie zugegangen werden, der genaue Zugangsweg wird jedoch durch den Operateur an Hand der MRI-Bilder und der genauen Lage des Tumors geplant und festgelegt.
Unterhalb des Schädelknochens folgt die Hirnhaut, auch Dura genannt, welche im nächsten Schritt eröffnet wird.
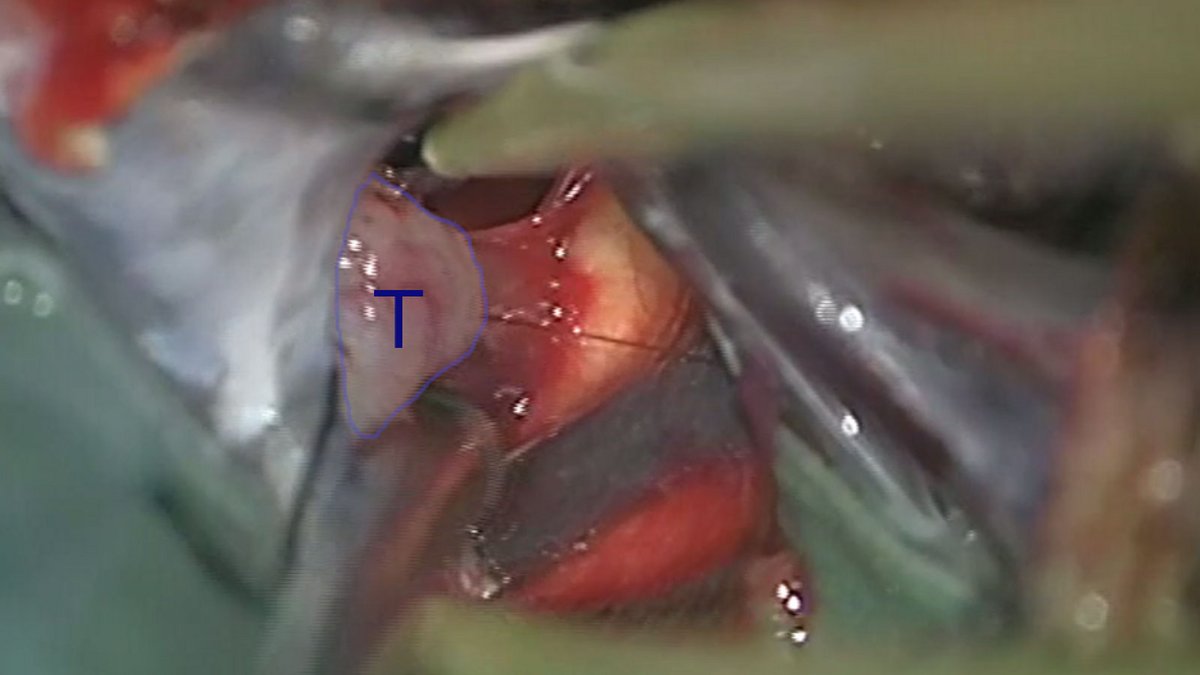
Unter mikroskopischer Kontrolle sowie mit Hilfe der Neuronavigation wird der Tumor möglichst gewebeschonend unter Berücksichtigung der umgebenden Strukturen freigelegt. Ist dies geschehen, erfolgt die Resektion. Das maximale Resektionsmass ist abhängig davon, inwieweit der Tumor das umliegende Gewebe infiltriert hat.
Nach gründlicher Blutstillung wird die Dura wieder dicht verschlossen.
Der vorher entnommene Schädelknochen wird mittels dünnen Titanplättchen und Schrauben wieder korrekt platziert und fixiert. Abschliessend wird die Wunde mit Klammern oder Nähten verschlossen.
Nach der Operation kommen die Patienten zur engmaschigen neurologischen Überwachung auf unsere Überwachungsstation. Bei einem normalen Verlauf können die Patienten üblicherweise am 1. oder 2. Tag nach der Operation zurück auf die Bettenstation verlegt werden. In diesem Zeitraum erfolgt ausserdem ein erneutes MRI zur postoperativen Kontrolle des Resektionsausmasses. Davon ist die weitere Therapieplanung abhängig *, *, *.
Behandlungsalternativen
Bei Patienten mit einem Hydrozephalus kann alternativ eine interne Liquorableitung über einen ventrikuloperitonealen Shunt erwogen werden. In Frage kommt eventuell auch eine endoskopische Drittventrikulozisternostomie, bei der eine Kurzschlussverbindung zwischen den inneren und äusseren Hirnwasserräumen hergestellt wird. Hier werden aber keine Proben der Zyste zur Untersuchung entnommen *, *, *, *.
Referenzen
-
Greenberg MS. Handbook of Neurosurgery. Thieme; 2016:1664.
-
Harbaugh R, Shaffrey CI, Couldwell WT. Neurosurgery Knowledge Update. Thieme; 2015:984.
-
Zaazoue MA, Goumnerova LC. Pineal region tumors: a simplified management scheme. [editorial]. Childs Nerv Syst 2016;32(11):2041.
-
Sonabend AM, Bowden S, Bruce JN. Microsurgical resection of pineal region tumors. J Neurooncol. 2016;130:351-366.
-
Májovský M, Netuka D, Beneš V. Clinical management of pineal cysts: a worldwide online survey. Acta Neurochir (Wien). 2016;158:663-669.