Meningeome sind langsam wachsende Tumoren, die von den Hirnhäuten ausgehen. Sie zählen mit rund 35 % zu den häufigsten Tumoren des zentralen Nervensystems (ZNS) und treten vermehrt ab dem 50. Lebensjahr auf. Mehr als 90 % der Meningeome sind gutartig. Wenn Meningeome kontinuierlich wachsen und eine Grösse erreichen, die das Hirn komprimiert und Symptome verursacht, sollten sie behandelt werden.
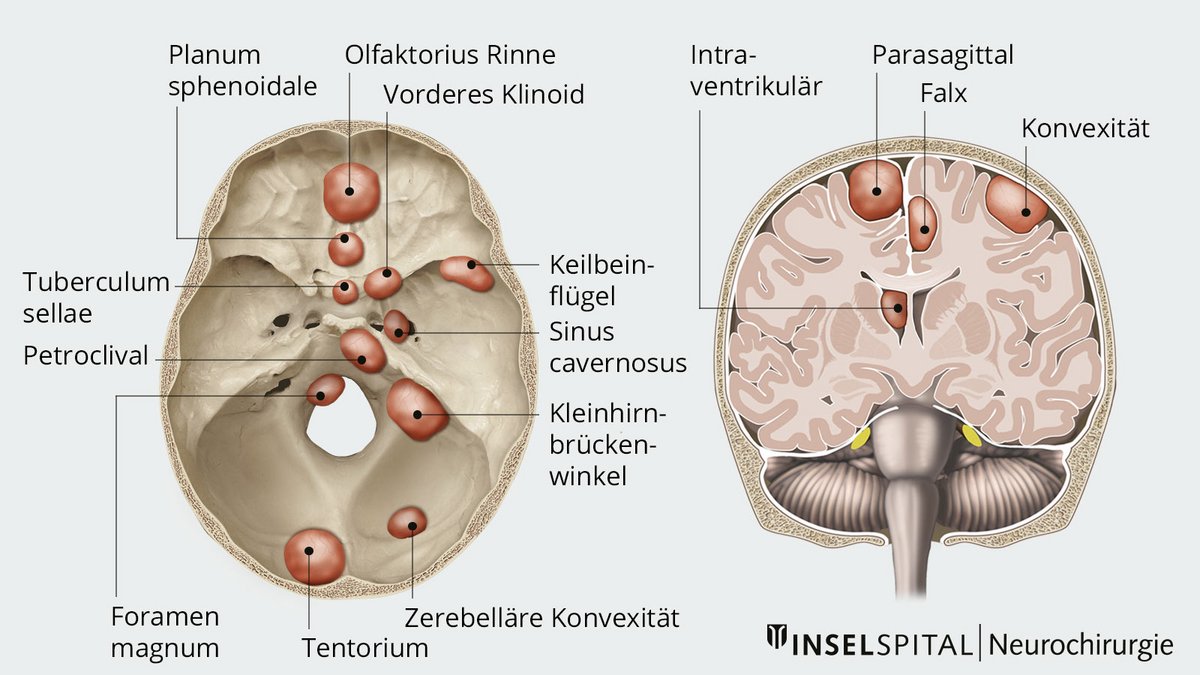
Was ist ein Meningeom?
Hirn und Rückenmark sind von drei schützenden Membranen umgeben, den sogenannten Meningen. Tumoren, die aus der Spinnwebenhaut (Arachnoidea) der Meningen entspringen, werden als Meningeome bezeichnet. Meningeome sind die häufigsten gutartigen intrakraniellen Tumoren.
Je nach Lage verursacht ein Meningeom unterschiedliche Beschwerden.
Meningeome wachsen in der Regel sehr langsam, können jedoch in seltenen Fällen das umgebende Gewebe infiltrieren und Metastasen bilden. Obwohl Meningeome meistens gutartig sind, bleiben sie mitunter aufgrund ihres schleichenden, verdrängenden Wachstums über einen längeren Zeitraum unerkannt und können so zu schweren Schäden und Behinderungen führen.
Was sind die Ursachen für Meningeome?
Ionisierende Strahlung gilt nach heutigem Wissensstand als der wichtigste bekannte Risikofaktor für die Entstehung von Meningeomen. Personen, die hohen Strahlendosen ausgesetzt waren, haben ein deutlich erhöhtes Risiko, im Laufe ihres Lebens ein Meningeom zu entwickeln. Dazu gehören beispielsweise Überlebende der Atombombenabwürfe, bei denen ein 6- bis 10-fach erhöhtes Risiko beschrieben wurde, sowie Patientinnen und Patienten nach einer Strahlentherapie im Kopf- und Halsbereich.
Auch Menschen mit der Erbkrankheit Neurofibromatose Typ 2 haben ein erhöhtes Risiko für die Entwicklung von Meningeomen.
Häufigkeit und Risikofaktoren
- Häufigste Tumoren des ZNS
- Grösster Risikofaktor: ionisierende Strahlung
- Vermehrtes Auftreten ab dem 50. Lebensjahr
Wie werden Meningeome eingeteilt?
Gemäss der World Health Organization (WHO) werden Meningeome anhand feingeweblicher (histopathologischer) Kriterien in drei Grade eingeteilt:
- WHO-Grad I: ca. 70–80 %
- WHO-Grad II: ca. 15–25 %
- WHO-Grad III: ca. 1–3 %
WHO-Grad-I-Meningeome sind die häufigste Form und in der Regel gutartig. Sie wachsen meist langsam und haben nach vollständiger Entfernung eine vergleichsweise geringe Rückfallrate.
WHO-Grad-II-Meningeome wachsen häufig schneller, können in das umgebende Gewebe einwachsen und weisen ein erhöhtes Risiko für ein Wiederauftreten (Rezidiv) auf.
WHO-Grad-III-Meningeome sind selten und bösartig (anaplastisch). Sie wachsen aggressiv, treten häufiger erneut auf und erfordern meist eine kombinierte Behandlung aus Operation und Strahlentherapie.
Welche Symptome verursachen Meningeome?
Meningeome wachsen in der Regel langsam. Durch ihr Wachstum können sie das umliegende Hirngewebe oder benachbarte Nerven und Gefässe verdrängen und dadurch Beschwerden verursachen. Welche Symptome auftreten, hängt vor allem von der Lage, der Grösse und dem Wachstumstempo des Tumors ab.
Da Meningeome meist langsam wachsen, entwickeln sich die Beschwerden häufig schleichend und nehmen im Verlauf allmählich zu. Nicht selten verursachen Meningeome über lange Zeit keine Beschwerden und werden zufällig im Rahmen einer Bildgebung entdeckt (Zufallsbefund).
Häufige Symptome:
- Kopfschmerzen
- epileptische Anfälle
- Seh-, Riech- oder Sprachstörungen
- Lähmungen oder Sensibilitätsstörungen
Was ist, wenn ein Meningeom zufällig entdeckt wird?
Viele Meningeome werden zufällig entdeckt, etwa im Rahmen einer Bildgebung wegen unspezifischer Kopfschmerzen oder nach einem Unfall. Solche Befunde bezeichnet man als Zufallsbefunde. Häufig erfolgt die Diagnose mittels Magnetresonanztomografie (MRT oder MRI) oder Computertomografie (CT).
Wenn das Meningeom klein ist, keine Beschwerden verursacht und nicht in der Nähe funktionell wichtiger Hirnareale liegt, wird in vielen Fällen zunächst eine regelmässige Verlaufsbeobachtung empfohlen. Diese erfolgt meist mittels wiederholter MRT-Kontrollen, häufig zunächst in jährlichen Abständen.
Bestimmte Merkmale können auf ein erhöhtes Wachstumspotenzial hinweisen. Dazu gehören unter anderem eine Tumorgrösse von mehr als 2,5 cm, ein begleitendes Hirnödem oder bestimmte Signalveränderungen in der T2-gewichteten MRT. In solchen Fällen – oder wenn der Tumor ungünstig gelegen ist – kann auch ein zufällig entdecktes Meningeom eine Behandlung erforderlich machen.
Wie werden Meningeome diagnostiziert?
Bildgebende Verfahren
Die Diagnose eines Meningeoms erfolgt meist im Rahmen der Abklärung neurologischer Symptome oder als Zufallsbefund bei einer Bildgebung aus anderen Gründen.
Sowohl in der Computertomografie (CT) als auch in der Magnetresonanztomografie (MRT oder MRI) zeigen sich Meningeome typischerweise als gut abgegrenzte, extraaxiale Tumoren, das heisst Tumoren, die primär ausserhalb des Hirngewebes entstehen. Charakteristisch ist ein breitflächiger Kontakt zur harten Hirnhaut (Dura mater) mit Verdrängung beziehungsweise Kompression des angrenzenden Hirngewebes.
Nach Gabe von Kontrastmittel zeigen Meningeome in der Regel eine homogene Kontrastmittelaufnahme. In der CT können zusätzlich Verkalkungen sowie sekundäre knöcherne Veränderungen nachgewiesen werden. In der MRT findet sich häufig eine nach aussen auslaufende durale Verdickung, das sogenannte Dural Tail Sign.

Wie werden Meningeome behandelt?
Kleine Meningeome, die nicht an Grösse zunehmen, können konservativ behandelt werden. Wachsen Meningeome kontinuierlich oder haben eine Grösse erreicht, die das Hirn komprimiert, ein Hirnödem verursacht oder symptomatisch wird, sollten sie behandelt werden. Standardtherapie ist die Operation, seltener eine radiochirurgische Therapie.
Aufgrund der variablen Lokalisation und Grösse muss die Entscheidung über das beste Vorgehen immer individuell besprochen werden. Die optimale Behandlung richtet sich also nach Grösse, Lage und Wachstumsgeschwindigkeit des Tumors, ausserdem nach der histopathologischen Gewebeanalyse sowie dem Allgemeinzustand des Patienten.
Konservative Behandlung
Patienten mit asymptomatischen Zufallsbefunden, kleineren Meningeomen oder höherem Lebensalter werden zunächst konservativ behandelt. Das bedeutet, es erfolgen regelmässig klinische Kontrollen inklusive Bildgebung (MRT, CT), um den Verlauf der Erkrankung zu überwachen. Die anfänglichen Nachkontrollen erfolgen innerhalb von 3–6 Monaten nach Stellen der Verdachtsdiagnose. Bei stabilen Befunden können die nachfolgenden Zeitintervalle zwischen den Kontrolluntersuchungen zunehmend verlängert werden.
In diversen Studien zeigte sich während der bildgebenden Verlaufskontrollen über einen durchschnittlichen Zeitraum von 2–5 Jahren bei rund 50–70 % der Patienten eine stabile Meningeomgrösse ohne Wachstumstendenz. Bei Grössenzunahme der Meningeome oder Entwicklung neuer Symptome sollte neu über eine weitere Therapie entschieden werden.
Operation
Die primäre Therapie für ein symptomatisches, sich vergrösserndes Meningeom besteht in einer vollständigen mikrochirurgischen Entfernung. Wenn dies möglich ist, bedeutet das in den meisten Fällen die Heilung für den Patienten.
Deshalb steht an oberster Stelle der Behandlung eine maximal mögliche, zugleich schonende und funktionserhaltende Tumorentfernung mit hohem kosmetischem Anspruch.
Dabei wird das Meningeom zuerst von innen heraus verkleinert, damit die Grenze zum Nachbargewebe entlastet wird. Danach wird der Tumor vorsichtig unter dem Mikroskop vom umgebenden normalen Gehirngewebe abpräpariert. Die tumortragende Hirnhaut wird meist mit entfernt und ersetzt. Die Verzahnung zwischen normalem Hirn und Meningeom ist dabei von Patient zu Patient unterschiedlich intensiv ausgeprägt. Ein Teil der Tumoren hat eine glatte Grenze, andere Tumoren haben die Hirnoberfläche infiltriert und werden von normalen Hirngefässen mit Blut versorgt.
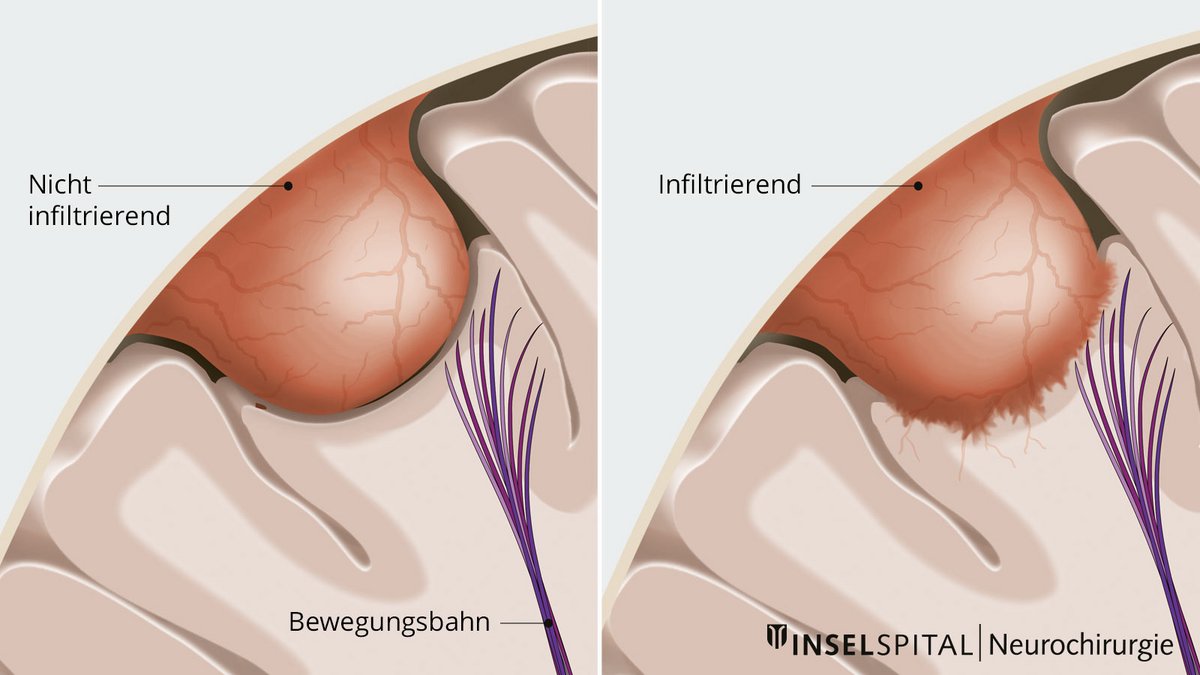
Wenn in diesen Fällen ein wichtiges Hirngebiet betroffen ist, muss die Operation unter Funktionsüberwachung durchgeführt werden. Dazu werden neueste technische Verfahren, wie die Neuronavigation mit Augmented Reality und das sogenannte intraoperative Neuromonitoring, eingesetzt, die dem Neurochirurgen ermöglichen, mit maximaler Präzision und Sicherheit unter dem Operationsmikroskop zu arbeiten und den Patienten gläsern zu machen, also in Gewebe hineinzuschauen.
Präoperative Embolisation
Meningeome sind oft stark durchblutet. Damit es während der Operation zu weniger Blutungen kommt, ist es wichtig, die Blutgefässe, die den Tumor versorgen, möglichst früh zu verschliessen.
In manchen Fällen – zum Beispiel bei bestimmten Meningeomen an der Schädelbasis – sind diese versorgenden Blutgefässe während der Operation nur schwer oder erst spät erreichbar. Dann kann vor dem eigentlichen Eingriff eine sogenannte endovaskuläre Embolisation durchgeführt werden. Dabei werden die Blutgefässe, die den Tumor versorgen, im Rahmen eines minimal-invasiven Gefässeingriffs gezielt verschlossen. So wird die Durchblutung des Tumors vermindert, was die Operation in ausgewählten Fällen sicherer machen und Blutungen reduzieren kann.
Bestrahlung
Bei Patienten mit einem behandlungsbedürftigen Meningeom, das nicht oder nur teilweise operierbar ist, ist die Strahlentherapie eine Behandlungsoption mit guter Tumorgrössenkontrolle. Die Strahlentherapie wird ausserdem unterstützend bei höhergradigen Meningeomen oder inkompletter Resektion eingesetzt. Die Meningeome dürfen dafür allerdings eine bestimmte Grösse nicht überschritten haben.
Radionuklidtherapie
In schwierigen Fällen mit progressivem Krankheitsfortschreiten trotz Operation und Bestrahlung bietet die Radionuklidtherapie eine weitere Therapiemöglichkeit. Dabei werden die Tumoren gezielt mit radioaktiven Medikamenten angegriffen. Das Radiopharmakon, die radioaktive Substanz, bindet an spezielle Rezeptoren auf der Oberfläche des Meningeoms, die Somatostatin-Rezeptoren, und erzielt dort eine lokale Strahlenwirkung und zerstört so die Tumorzellen.
Risiken des Eingriffs
Das Risiko von Komplikationen bei der operativen Entfernung eines Meningeoms – und gegebenenfalls bei einer zusätzlichen Strahlentherapie – hängt von verschiedenen Faktoren ab. Dazu zählen vor allem die Grösse, die Lage und die Zugänglichkeit des Tumors sowie das Alter und der allgemeine Gesundheitszustand der Patientin oder des Patienten.
Bei unkompliziert gelegenen Meningeomen ist das Risiko für schwere Komplikationen in der Regel gering und liegt unter 2 %. Bei komplex gelegenen oder grösseren Tumoren kann das Risiko höher sein.
Welche Risiken im individuellen Fall bestehen, hängt stark von der jeweiligen Situation ab. Gerne besprechen wir diese mit Ihnen persönlich anhand Ihrer Magnetresonanztomografie (MRT) und erklären Ihnen Ihr persönliches Risikoprofil.
Wie geht es nach der Operation weiter?
Die Prognose bei einem Meningeom hängt von mehreren Faktoren ab. Entscheidend sind vor allem die Lage und Grösse des Tumors sowie seine feingeweblichen Eigenschaften, also der WHO-Grad (I–III). Ein weiterer wichtiger Faktor ist, ob der Tumor bei der Operation vollständig oder nur teilweise entfernt werden konnte. Auch die Ergebnisse der feingeweblichen Untersuchung und – in einzelnen Fällen – molekulare Biomarker spielen bei der weiteren Behandlungsplanung eine Rolle. Auf Grundlage dieser Befunde wird das weitere Vorgehen individuell festgelegt.
WHO-Grad I und Totalresektion
Eine Kontrolle nach der Operation, je nach Risiko eine weitere Kontrolle nach 2,5 und 10 Jahren
WHO-Grad I und unvollständige Resektion
Verlaufskontrollen jährlich und/oder Radiochirurgie
WHO-Grad II und Totalresektion
Verlaufskontrollen mittels MRI alle 6–24 Monate, ggf. Nachbestrahlung
WHO-Grad II und unvollständige Resektion
Verlaufskontrollen jährlich und/oder Radiochirurgie
WHO-Grad III
Nachbestrahlung, ggf. Radionuklidtherapie oder experimentelle Chemotherapie, sowie MRI-Verlaufskontrollen alle 3–6 Monate
Warum Sie sich am Inselspital behandeln lassen sollten
Am Inselspital wird für jede Patientin und jeden Patienten eine individuell abgestimmte Behandlungsstrategie festgelegt. Dies erfolgt im zertifizierten Hirntumorzentrum, wo ein interdisziplinäres Team gemeinsam die verschiedenen Behandlungsmöglichkeiten prüft und die bestmögliche Therapie empfiehlt.
Die Besprechung findet im Rahmen eines wöchentlichen Tumorboards statt. Dort beraten Spezialistinnen und Spezialisten aus den Bereichen Neurochirurgie, Neurologie, Neuroonkologie, Nuklearmedizin, Radioonkologie und Pathologie gemeinsam über jeden einzelnen Fall.
So stellen wir sicher, dass alle wichtigen fachlichen Perspektiven in die Behandlungsentscheidung einfliessen.