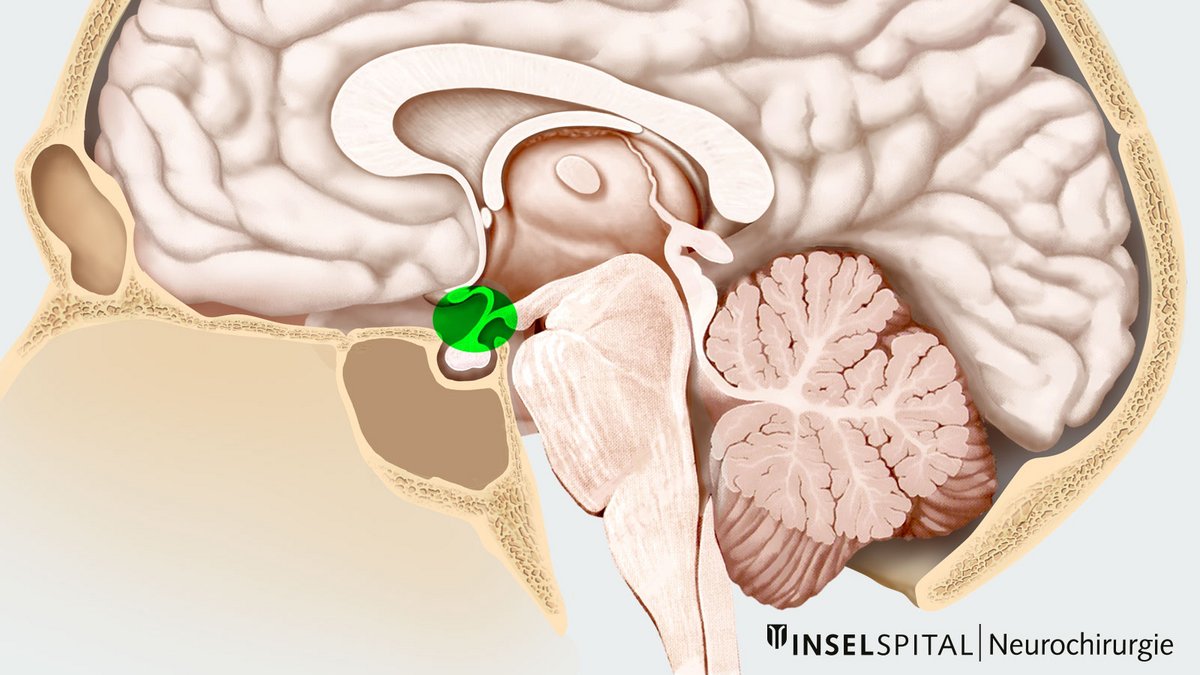
Kraniopharyngeome sind Tumoren, die meistens im Bereich der Hirnanhangsdrüse (Hypophyse) entstehen. Ein Kraniopharyngeom entwickelt sich aus Zellresten aus der Embryonalzeit und tritt deshalb häufig bei Kindern auf. Kraniopharyngeome sind zwar gutartig und wachsen langsam, können aber stark mit den umgebenden Strukturen wie Hypophyse, Hypophysenstiel, Sehnerven und Gefässen verwachsen.
Wie häufig ist ein Kraniopharyngeom und wer ist betroffen?
Kraniopharyngeome machen etwa 2,5–4 % aller Hirntumoren aus. Etwa die Hälfte der Kraniopharyngeome treten im Kindesalter auf, mehrheitlich zwischen dem 5. und 10. Lebensjahr. Die andere Hälfte dieser Tumoren findet sich bei Erwachsenen, meistens ab dem 40. Lebensjahr. Frauen und Männer sind gleich häufig betroffen.
Wie entsteht ein Kraniopharyngeom?
Kraniopharyngeome entstehen, wenn sich Restzellen der Rathke-Tasche plötzlich vermehren. Die Rathke-Tasche ist eine Struktur aus der Embryonalentwicklung der Hypophyse, die sich normalerweise zurückbildet.
Kraniopharyngeome entstehen meistens im Bereich des Hypophysenstiels und können sich von dort in verschiedene Richtungen (suprasellär, parasellär oder sellär) ausdehnen. Eine Minderheit an Kraniopharyngeomen (etwa 5 %) wachsen rein intraventrikulär (im 3. Ventrikel).
Mediziner unterscheiden zwei Formen von Kraniopharyngeomen aufgrund ihrer feingeweblichen Struktur:
- Das adamantinöse Kraniopharyngeom tritt häufiger bei Kindern auf, ist oft hart und verkalkt und hat ein höheres Rezidivrisiko.
- Das papilläre Kraniopharyngeom kommt hauptsächlich bei Erwachsenen vor, ist nur äusserst selten verkalkt und hat ein geringeres Rezidivrisiko.
Welche Symptome verursacht ein Kraniopharyngeom?
Aufgrund der unmittelbaren Nähe zu den Sehnerven, der Kreuzung der Sehnerven, dem sogenannten Chiasma opticum, der Hypophyse, dem Hypophysenstiel, dem Hypothalamus (Teil des Zwischenhirns) und dem Hirnstamm kann es zu einer ganzen Bandbreite an Symptomen kommen:
- Kopfschmerzen
- Sehstörungen und/oder Gesichtsfeldeinschränkungen
- hormonelle Störungen, insbesondere Diabetes insipidus (Trink- und Ausscheidungsmenge erhöht, Durst)
- Übelkeit und Erbrechen
- neurokognitive Defizite
- hypothalamische Störungen, insbesondere Gewichtszunahme
Wie wird ein Kraniopharyngeom diagnostiziert?
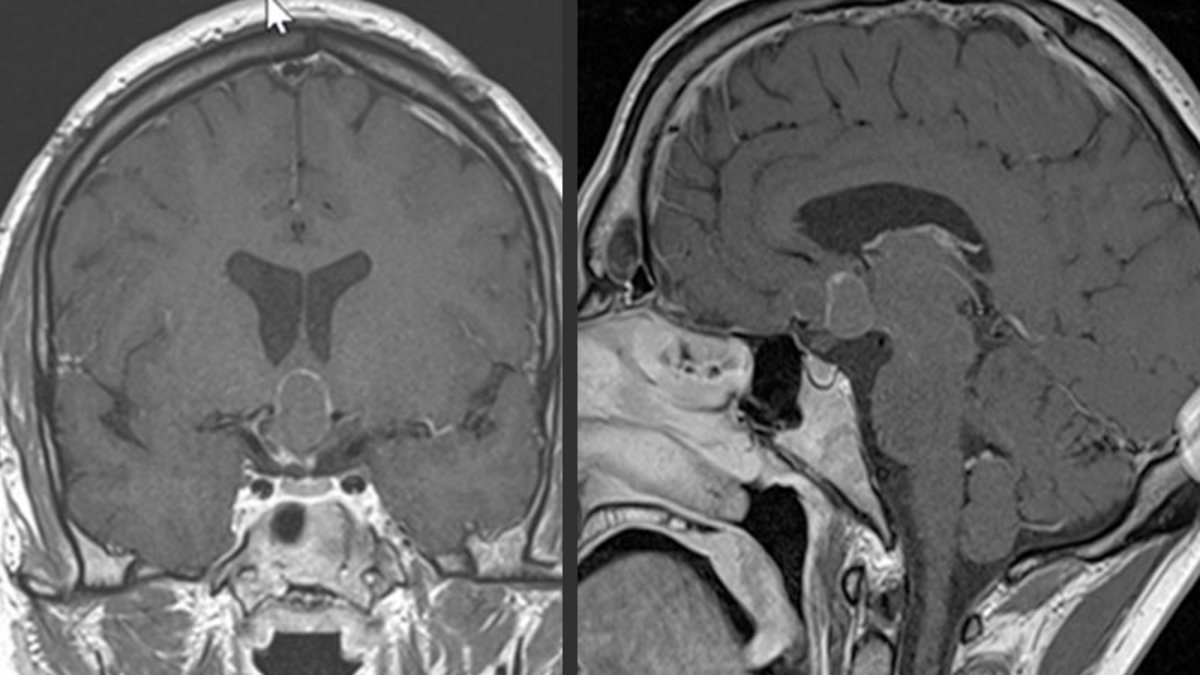
Eine Magnetresonanztomografie (MRT bzw. MRI von engl. Magnetic Resonance Imaging) mit Kontrastmittel ist das diagnostische Verfahren der ersten Wahl. Das MRI-Bild gibt Informationen über Lokalisation und Ausdehnung des Tumors.
Manchmal ist eine ergänzende Computertomografie (CT) hilfreich, um das Vorhandensein von Verkalkungen zu beurteilen.
Bei allen Patienten mit Verdacht auf ein Kraniopharyngeom ist eine endokrinologische Abklärung empfohlen, um mögliche hormonelle Störungen zu diagnostizieren und zu behandeln.
Eine augenärztliche Untersuchung wird notwendig, wenn das MRI-Bild zeigt, dass der Tumor Kontakt zum Chiasma opticum hat, der Stelle, an der sich die Sehnerven der Augen kreuzen.
Wie wird ein Kraniopharyngeom behandelt?
Durch ihr verdrängendes Wachstum können die Tumoren zu einer dauerhaften Schädigung der umgebenden Hirnstrukturen führen. Die Therapie der Wahl ist deshalb die vollständige chirurgische Entfernung des Tumors. Wenn aufgrund der Nähe zu wichtigen umliegenden Strukturen eine vollständige Resektion nicht möglich ist, sollte eine begleitende Radiotherapie bzw. Radiochirurgie diskutiert werden, um das Rezidivrisiko zu senken.
Die medizinische Forschung arbeitet derzeit auch an der Entwicklung von Chemotherapeutika als ergänzende Therapie. Aktuell werden Medikamente für die Therapie des papillären Kraniopharyngeoms getestet. Es handelt sich dabei um sogenannte BRAF-Inhibitoren wie Vemurafenib und Dabrafenib.
Es gibt zwei chirurgische Zugangswege für die Operation eines Kraniopharyngeoms:
- der transkranielle Zugang
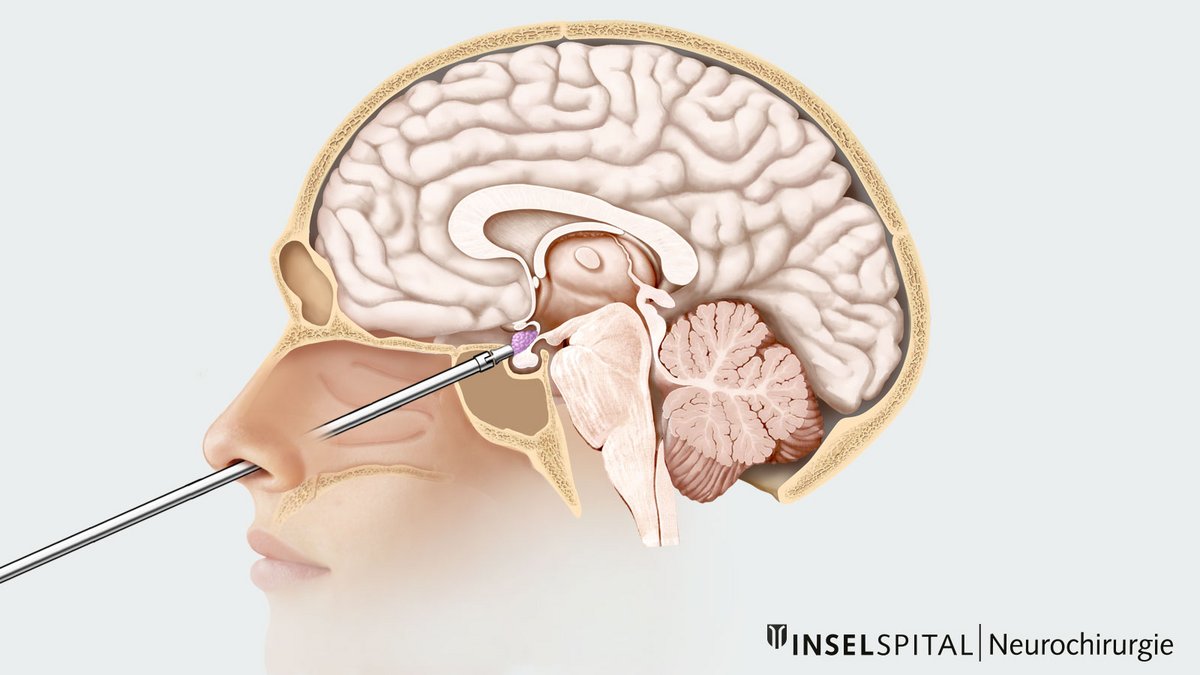
- der transnasale transsphenoidale Zugang
Bei klassischen Kraniopharyngeomen können sowohl der transkranielle wie auch der transnasale transsphenoidale Zugang gewählt werden.
Beim transkraniellen Zugang wird eine Öffnung im Schädel vorgenommen, eine sogenannte Kraniotomie. Der Tumor kann danach mikrochirurgisch entfernt werden. Dies ist die empfohlene Technik, insbesondere bei rein intraventrikulären Kraniopharyngeomen, die vollständig oder teilweise in den Hirnkammern (Ventrikel) liegen, oder bei Tumoren, die ein ausgedehntes Wachstum zeigen.
Beim transnasalen transsphenoidalen Zugang erfolgt der operative Zugangsweg durch die Nase.
Mögliche Komplikationen nach Resektion eines Kraniopharyngeoms sind hauptsächlich Störungen des Elektrolyt- und des Hormonhaushalts. Diese Störungen können vorübergehender Natur oder auch permanent sein. In der postoperativen Phase werden die Blutwerte und der Wasserhaushalt engmaschig kontrolliert. Gegebenenfalls muss ein Hormonmangel medikamentös ersetzt werden.
Warum Sie sich am Inselspital behandeln lassen sollten
Unser spezialisiertes neurochirurgisches Team verfügt über eine langjährige chirurgische Erfahrung bei der Operation von Kraniopharyngeomen. Abhängig von Lokalisation und Ausdehnung des Tumors bieten wir unseren Patientinnen und Patienten die für sie sicherste und effektivste Operationstechnik an.
Die Neurochirurgie am Inselspital arbeitet dabei eng mit den Fachabteilungen der Endokrinologie, Ophthalmologie und Radio-Onkologie zusammen. So können wir Ihnen eine umfassende und qualitativ hochstehende Betreuung gewährleisten – vor, während und nach der Operation.
Ausserdem wird jeder Fall im Rahmen unseres interdisziplinären Hypophysen-Boards und des neuroonkologischen Tumorboards besprochen, wo Neurochirurgen zusammen mit Endokrinologen, Onkologen und Radioonkologen einen auf jeden Patienten abgestimmten, individuellen Therapieplan erarbeiten.
Weiterführende Literatur
- Greenberg MS. Handbook of Neurosurgery. Thieme; 2016:1664.
- Komotar RJ, Starke RM, Raper DMS, Anand VK, Schwartz TH. Endoscopic endonasal compared with microscopic transsphenoidal and open transcranial resection of craniopharyngiomas. World neurosurgery. 2012;77:329-341.
- Liubinas SV, Munshey AS, Kaye AH. Management of recurrent craniopharyngioma. Journal of Clinical Neuroscience. 2011;18:451-457.
- Minamida Y, Mikami T, Hashi K, Houkin K. Surgical management of the recurrence and regrowth of craniopharyngiomas. Journal of neurosurgery. 2005
- Moussazadeh N, Prabhu V, Bander ED et al. Endoscopic endonasal versus open transcranial resection of craniopharyngiomas: a case-matched single-institution analysis. Neurosurgical Focus. 2016;41:E7.
- Müller HL. Craniopharyngioma. Endocrine reviews. 2014;35:513-543.
- Ottenhausen M, Rumalla K, La… EC. Treatment strategies for craniopharyngiomas. Journal of Neurosurgical Sciences. 2017