Das pilozytische Astrozytom ist der häufigste gutartige Hirntumor bei Kindern und Jugendlichen. Durch eine mikroneurochirurgische Behandlung ist er meistens heilbar, selbst wenn er sich an einer schwierig zu operierenden Stelle im Gehirn befindet. Nach der vollständigen Entfernung des Tumors sind eine Bestrahlung oder Chemotherapie nicht notwendig. Neben Informationen zur Standardtherapie erhalten Sie nachfolgend auch Informationen über Spezialfälle, wie beispielsweise die Behandlung von Kindern mit Neurofibromatose.
Was sind pilozytische Astrozytome?
Pilozytische Astrozytome sind die häufigsten gutartigen Hirntumoren des Kindes und jungen Erwachsenen und machen ca. 5 % aller Gliome aus *. Sie entstehen aus den Astrozyten, die zum Stützgewebe (Gliazellen) des Zentralnervensystems gehören und werden deshalb den Gliomen zugeordnet. Pilozytische Astrozytome weisen mikroskopisch einen faserartigen «pilozytären» Charakter auf. Makroskopisch sind sie häufig zystisch, das heisst die pilozytischen Astrozytome bestehen aus einem Gemisch von Flüssigkeit gefüllten Hohlräumen und festen Anteilen. Die meisten pilozytischen Astrozytome sind langsam wachsende Tumoren, die sich nicht in andere Körperregionen ausbreiten und eine sehr gute Prognose aufweisen.
Wo wachsen pilozytische Astrozytome?
Bei Kindern gehen pilozytische Astrozytome besonders häufig aus den Astrozytenzellen des Kleinhirns hervor, einer Region, die für Bewegung und Gleichgewicht eine entscheidende Rolle spielt. Pilozytische Astrozytome können jedoch auch im Bereich der Sehbahn, des Grosshirns oder im Hirnstamm auftreten *, *.
Wie häufig sind pilozytische Astrozytome?
Pilozytische Astrozytome sind die häufigsten primären, gutartigen Hirntumoren bei Kindern im Alter von 0–14 Jahren. Sie können aber auch noch bis ins Erwachsenenalter auftreten und kommen bei Männern geringfügig häufiger vor als bei Frauen (im Verhältnis 1,1 : 1). Pilozytische Astrozytome treten bei Kindern mit einer Häufigkeit von 0,8 pro 100 000 Personen pro Jahr auf.
Welche Symptome verursacht ein pilozytisches Astrozytom?
Pilozytische Astrozytome können durch ihr Wachstum den Druck im Inneren des Schädels erhöhen und führen dann zu:
- morgendlichen Kopfschmerzen
- Übelkeit und Erbrechen
- Müdigkeit
Darüber hinaus können je nach Lage des Tumors weitere Symptome auftreten:
- Gang- und Koordinationsstörungen
- Doppelbilder
- Sehstörungen
- zunehmende Somnolenz (Schläfrigkeit, Benommenheit)
- in seltenen Fällen epileptische Anfälle
- Störungen von Bewegung und Sprache
- schleichende Wechsel von Stimmung und Persönlichkeit
Aufgrund des meist sehr langsamen Wachstums der pilozytischen Astrozytome können die Symptome schleichend auftreten und über längere Zeit unbemerkt bleiben.
Warum entstehen pilozytische Astrozytome?
Die Ursachen für die Entstehung pilozytischer Astrozytome sind aktuell Gegenstand intensiver Forschung. Bei einem Grossteil dieser Tumoren findet sich eine spezielle Variante eines Gens namens BRAF, das an der Steuerung der Zellteilung beteiligt ist *. Bei pilozytischen Astrozytomen weist das BRAF-Gen eine Mutation auf, die eine unkontrollierte Teilung der Tumorzellen initiiert. Der Effekt wird über den sogenannten Mitogen-aktivierten Proteinkinase-Weg (MAP-Kinase-Weg oder MAPK-Weg) vermittelt *. Dieser ist auch mit Genen der Zellalterung assoziiert, wodurch sich das langsame Wachstum und die in seltenen Fällen beschriebene Spontanregression pilozytischer Astrozytome erklären lässt *, *.
Wie werden pilozytische Astrozytome diagnostiziert?
Bildgebende Verfahren spielen eine Schlüsselrolle bei der Diagnose von pilozytischen Astrozytomen. Dabei eignet sich die Magnetresonanztomografie (MRT bzw. MRI von engl. Magnetic Resonance Imaging) in Kombination mit einem intravenösen Kontrastmittel besonders gut, um pilozytische Astrozytome zu visualisieren und vom umgebenden gesunden Hirngewebe abzugrenzen. Per MRI lässt sich jedoch nicht mit 100%iger Sicherheit ein pilozytisches Astrozytom diagnostizieren oder ausschliessen, dass es sich um einen bösartigeren Tumor handelt. Dies ist erst nach einer Gewebeentnahme und feingeweblicher mikroskopischer Untersuchung möglich. Dieser Prozess nimmt mehrere Tage in Anspruch. Die Gewebeentnahme erfolgt entweder als Biopsie oder häufiger gleich bei der chirurgischen Entfernung des pilozytischen Astrozytoms.
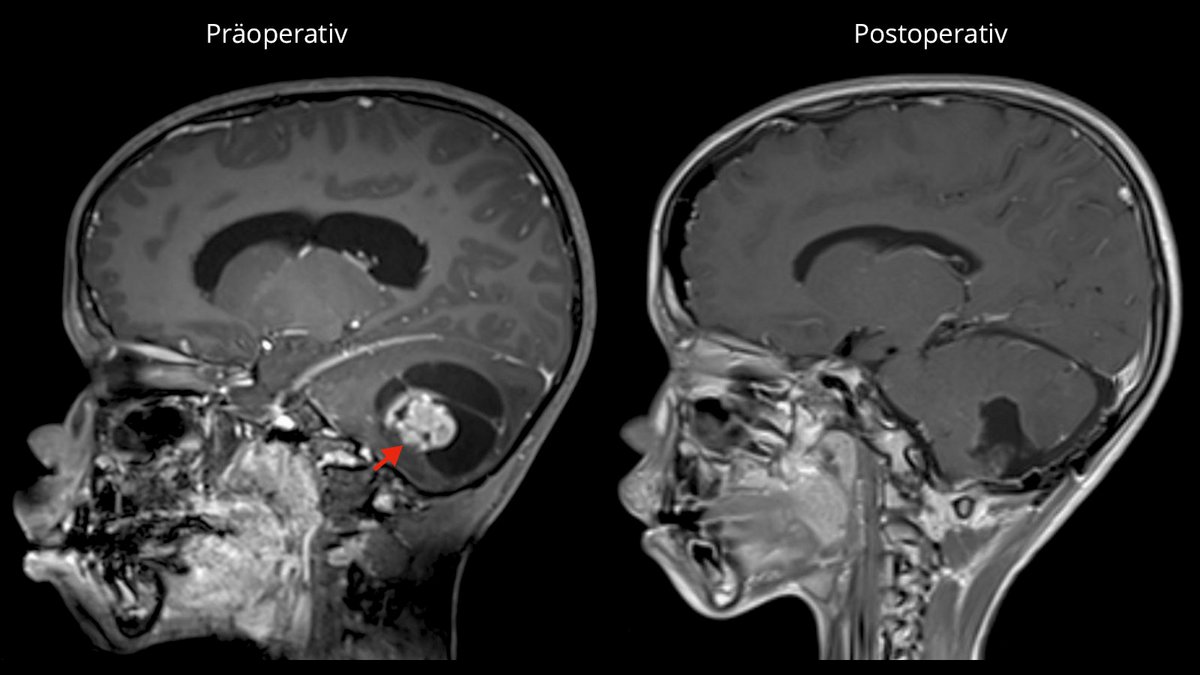
Wie sieht die Behandlung eines pilozytischen Astrozytoms aus?
Die Operation als Therapie der ersten Wahl

Die Behandlung erster Wahl ist, sofern möglich, die komplette mikrochirurgische Entfernung des pilozytischen Astrozytoms. Insbesondere im jugendlichen Alter kann nach vollständiger Entfernung des Tumors in den meisten Fällen eine Heilung erreicht werden mit einer 10-Jahres-Überlebensrate von bis zu 100 % und einer Rezidivrate von nur 2–5 % *. Nach einer vollständigen Tumorentfernung sind auch keine Bestrahlung oder Chemotherapie notwendig.
In einigen Fällen ist es allerdings nicht möglich, ein pilozytisches Astrozytom vollständig zu entfernen, da dieses in lebenswichtige Strukturen wie den Hirnstamm einwächst. Sofern der Tumor eine klare Grenze zum umliegenden Hirngewebe hat, kann auch in diesen Fällen eine Teilentfernung des Tumors sinnvoll sein.
Obschon es im Fall eines Resttumors bei etwa der Hälfte der Patienten innerhalb von 5 Jahren zu einem Fortschreiten des Tumors kommt, beträgt das 5-Jahres-Überleben dennoch 90 % *, *, *. Die Komplikationsrate einer chirurgischen Tumorentfernung und einer eventuellen weiteren Behandlung mittels Radio- und/oder Chemotherapie wird durch etliche Faktoren wie Grösse und Lage des Tumors, dessen Zugänglichkeit sowie den klinischen Zustand und das Alter des Patienten beeinflusst.
Die Strahlentherapie als Ergänzung bei Tumorresten
Falls nach einer Operation ein Rest eines pilozytischen Astrozytoms verbleibt und dieser nicht chirurgisch entfernt werden kann, stellt die Strahlentherapie eine wirkungsvolle Ergänzung dar. In den letzten Jahren hat insbesondere die Radiochirurgie, eine sehr gezielte und intensive Art der Bestrahlung, zunehmend an Bedeutung gewonnen. Mittels Radiochirurgie kann sowohl bei einem kleinen Resttumor als auch bei einem Rezidiv eine gute Tumorkontrolle erreicht werden *, *, *. Alternativ kann man im Fall von Resttumor mit einer weiteren Therapie auch zuwarten und den Patienten so lange engmaschig kontrollieren, bis der Tumorrest weiterwächst.
Vermeidung der Strahlentherapie bei jüngeren Kindern
Eine Bestrahlung im frühen Kindesalter kann zu kognitiven und hormonellen Problemen sowie zu Gefässveränderungen führen *, *. Aus diesem Grund versucht man bei jüngeren Kindern unter 5–10 Jahren, die Bestrahlung hinauszuzögern und eventuelle Tumorreste nach einer Operation durch eine Chemotherapie zu bekämpfen, um dem wachsenden Hirn den potenziell schädlichen Effekt der Bestrahlung zu ersparen *, *.
Spezialfall Neurofibromatose Typ 1
In einigen Fällen sind pilozytische Astrozytome auf eine Erbkrankheit, die Neurofibromatose Typ 1 (NF1), zurückzuführen. In diesem Fall treten die Tumoren mit einem anderen Verteilungsmuster auf und wachsen in der Regel auch noch langsamer.
Bei Patienten mit Neurofibromatose Typ 1 treten pilozytische Astrozytome am häufigsten (zu zwei Drittel) im Bereich des Sehnervs oder der Sehbahn, zu 20 % im Hirnstamm und nur selten (zu 5 %) im Kleinhirn auf *. Diese Tumoren weisen ein anderes Wachstumsverhalten auf und erfordern eine angepasste Therapie. NF1-assozierte Tumoren weisen eine bessere Prognose auf als sporadische Gliome. Es existieren sogar Berichte von Spontanregressionen, was auf die oben genannte Aktivierung der Mechanismen der Zellalterung zurückgeführt wird *.
Gliome der Sehbahn
Bei Kindern mit Neurofibromatose Typ 1 werden Gliome der Sehbahn in 15–20 % der Fälle entdeckt, wovon jedoch nur die Hälfte dieser Patienten jemals Beschwerden erleidet *. Bei mehr als der Hälfte der Tumoren der Sehbahn handelt es sich nach feingeweblicher Untersuchung um pilozytische Astrozytome *. Für Gliome der Sehbahn ist in den meisten Fällen keine Behandlung notwendig *. Sollte eine Behandlung irgendwann erforderlich werden, erfolgt diese primär durch eine Chemotherapie.
Spezialfall Pilomyxoides Astrozytom
Verwandt mit den pilozytischen Astrozytomen sind die pilomyxoiden Astrozytome. Sie teilen zwar viele genetische Eigenschaften mit pilozytischen Astrozytomen, bilden jedoch eine eigene Untergruppe mit einem aggressiveren Verhalten *. Während pilozytische Astrozytome nach WHO als Grad-I-Tumoren eingeteilt werden, erhalten die pilomyxoiden Astrozytome in der neuen WHO-Klassifikation von 2016 keine Gradeinteilung mehr (vormals Grad II) *.
Warum Sie sich am Inselspital behandeln lassen sollten
Am Inselspital sind wir stets bestrebt, für unsere Patientinnen und Patienten die individuell bestmögliche Behandlungsstrategie zu finden. Dies gelingt uns am zertifizierten neuroonkologischen Tumorzentrum durch enge interdisziplinäre Zusammenarbeit.
In wöchentlich stattfindenden Tumor Boards besprechen Spezialistinnen und Spezialisten aus Neurochirurgie, Neurologie, Neuroonkologie, Nuklearmedizin, Radioonkologie und Pathologie jede Patientin und jeden Patienten individuell, um die optimale Behandlung festzulegen.
Für die operative Behandlung nutzen wir innovative technische Verfahren wie die Neuronavigation und das sogenannte intraoperative Neuromonitoring. Diese ermöglichen maximale Präzision und höchste Sicherheit für unsere Patientinnen und Patienten.
Referenzen
-
Ostrom QT, Gittleman H, Xu J et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009-2013. Neuro Oncol. 2016;18:v1-v75.
-
Fernandez C, Figarella-Branger D, Girard N et al. Pilocytic astrocytomas in children: prognostic factors--a retrospective study of 80 cases. Neurosurgery. 2003;53:544-53; discussion 554.
-
Bond KM, Hughes JD, Porter AL, Orina J, Fang S, Parney IF. Adult Pilocytic Astrocytoma: An Institutional Series and Systematic Literature Review for Extent of Resection and Recurrence. World Neurosurg. 2018;110:276-283.
-
Korshunov A, Meyer J, Capper D et al. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol. 2009;118:401-405.
-
Tatevossian RG, Lawson AR, Forshew T, Hindley GF, Ellison DW, Sheer D. MAPK pathway activation and the origins of pediatric low-grade astrocytomas. J Cell Physiol. 2010;222:509-514.
-
Jacob K, Quang-Khuong DA, Jones DT et al. Genetic aberrations leading to MAPK pathway activation mediate oncogene-induced senescence in sporadic pilocytic astrocytomas. Clin Cancer Res. 2011;17:4650-4660.
-
Raabe EH, Lim KS, Kim JM et al. BRAF activation induces transformation and then senescence in human neural stem cells: a pilocytic astrocytoma model. Clin Cancer Res. 2011;17:3590-3599.
-
Trifiletti DM, Peach MS, Xu Z, Kersh R, Showalter TN, Sheehan JP. Evaluation of outcomes after stereotactic radiosurgery for pilocytic astrocytoma. J Neurooncol. 2017;134:297-302.
-
Simonova G, Kozubikova P, Liscak R, Novotny J. Leksell Gamma Knife treatment for pilocytic astrocytomas: long-term results. J Neurosurg Pediatr. 2016;18:58-64.
-
Kano H, Niranjan A, Kondziolka D et al. Stereotactic radiosurgery for pilocytic astrocytomas part 2: outcomes in pediatric patients. J Neurooncol. 2009;95:219-229.
-
Ater JL, Zhou T, Holmes E et al. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30:2641-2647.
-
Gnekow AK, Walker DA, Kandels D et al. A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma - A final report. Eur J Cancer. 2017;81:206-225.
-
Merchant TE, Kun LE, Wu S, Xiong X, Sanford RA, Boop FA. Phase II trial of conformal radiation therapy for pediatric low-grade glioma. J Clin Oncol. 2009;27:3598-3604.
-
Merchant TE, Conklin HM, Wu S, Lustig RH, Xiong X. Late effects of conformal radiation therapy for pediatric patients with low-grade glioma: prospective evaluation of cognitive, endocrine, and hearing deficits. J Clin Oncol. 2009;27:3691-3697.
-
Campen CJ, Gutmann DH. Optic Pathway Gliomas in Neurofibromatosis Type 1. J Child Neurol. 2018;33:73-81.
-
Rodriguez FJ, Perry A, Gutmann DH et al. Gliomas in neurofibromatosis type 1: a clinicopathologic study of 100 patients. J Neuropathol Exp Neurol. 2008;67:240-249.
-
Rubin JB, Gutmann DH. Neurofibromatosis type 1 - a model for nervous system tumour formation. Nat Rev Cancer. 2005;5:557-564.
-
Banan R, Hartmann C. The new WHO 2016 classification of brain tumors-what neurosurgeons need to know. Acta Neurochir (Wien). 2017;159:403-418.