Ependymome sind meist langsam wachsende Tumoren des zentralen Nervensystems. Sie können grundsätzlich in jedem Lebensalter auftreten, sind aber bei Kindern und Jugendlichen häufiger. Während einige Ependymome völlig symptomlos bleiben, können andere schwerwiegende neurologische Probleme verursachen, insbesondere dann, wenn sie den Abfluss des Hirnwassers blockieren. Die optimale Therapie besteht in der kompletten chirurgischen Entfernung, wobei in vielen Fällen zusätzlich eine Strahlen- oder Chemotherapie erforderlich sind, um das Risiko eines Rückfalls zu minimieren.
Wer ist von Ependymomen betroffen?
Ependymome können prinzipiell in jeder Altersgruppe und im gesamten Bereich des Ventrikelsystems sowie des Rückenmarks auftreten. Die Tumorlokalisation und das Erkrankungsalter variieren dabei je nach Ependymom-Typ.
Grundsätzlich gilt:
- Ependymome werden häufiger im Kindes- und Jugendalter diagnostiziert, wobei etwa 25 % bei Kindern unter 3 Jahren auftreten.
- Ependymome machen etwa 10 % aller pädiatrischen Hirntumoren aus.
- Fast zwei Drittel der Ependymome im Schädel treten bei Kindern auf, während Tumoren im Rückenmark fast ausschließlich bei Erwachsenen oder Jugendlichen diagnostiziert werden.
- Neben dem häufigen Auftreten bei Kindern zeigen sich Ependymome bei Erwachsenen vermehrt zwischen dem 30. und 60. Lebensjahr.
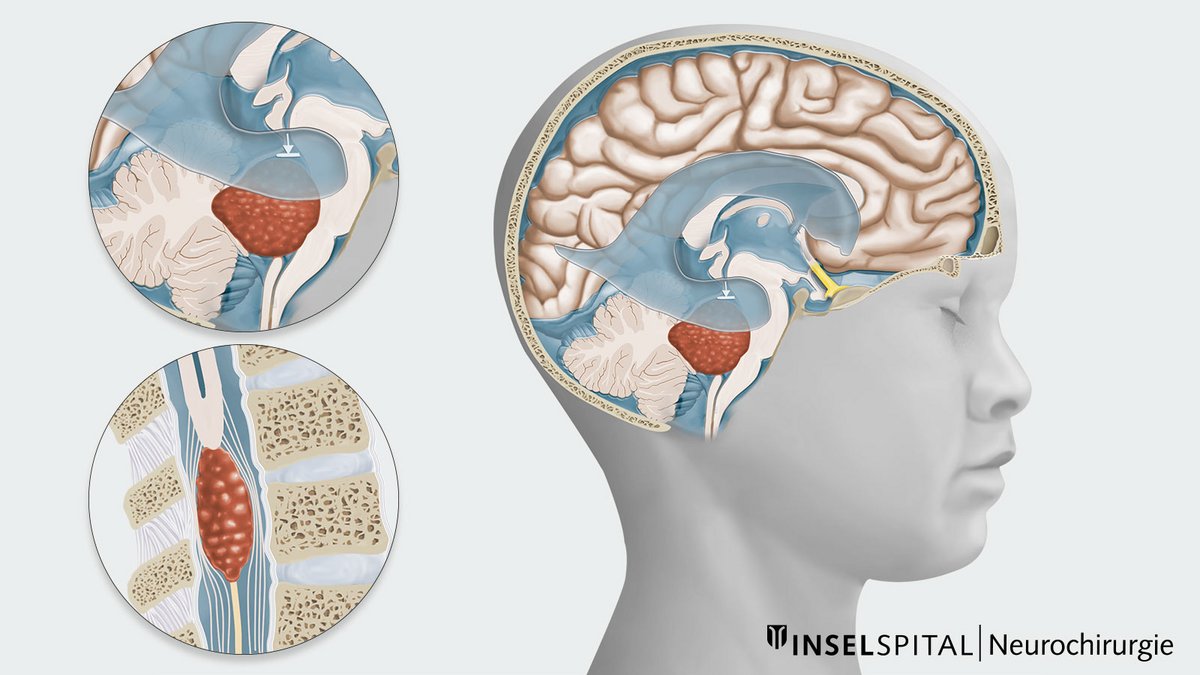
Welche Arten von Ependymomen gibt es?
Die Gruppe der Ependymome ist sehr heterogen und umfasst sowohl gutartige, langsam wachsende Tumoren als auch aggressive, schnell wachsende Tumoren. Zudem existieren verschiedene molekulare Subgruppen, die mit unterschiedlicher Prognose einhergehen.
Traditionell werden Ependymome nach ihrer Histologie in die WHO-Grade 1–3 unterteilt. Jedoch hat sich gezeigt, dass eine Risikoabschätzung anhand der molekularen Untergruppen, der Tumorlokalisation und des Patientenalters besser geeignet ist. Es liegen bisher noch nicht ausreichend Daten vor, um die einzelnen molekularen Subgruppen den WHO-Graden zuzuordnen *.
Gemäss der neuen WHO-Klassifikation werden Ependymome nun eingeteilt in *:
Supratentorielles Ependymom
Lokalisation Grosshirn, WHO-Grad 2–3, typischerweise bei jüngeren Kindern
Molekulare Subgruppen:
- ST-Ependymom mit ZFTA-Fusion (ehemals RELA-Fusion)
- ST-Ependymom mit YAP1-Fusion
- andere seltene Fusionen
Infratentorielles Ependymom
Lokalisation Posterior Fossa (hintere Schädelgrube), WHO-Grad 2–3, häufiger bei älteren Kindern und Erwachsenen
Molekulare Subgruppen:
- PFA-Ependymom (PF-Typ A)
- PFB-Ependymom (PF-Typ B)
Spinales Ependymom
MYCN-Amplifikation, WHO-Grad 2–3
Myxopapilläres Ependymom
früher WHO-Grad 1, jetzt WHO-Grad 2, spezielle Art von spinalen Ependymomen, meist bei älteren Kindern und jungen Erwachsenen
Subependymom
WHO-Grad 1
Welche Symptome verursacht ein Ependymom?
Im Gehirn wachsende Ependymome können je nach ihrer Wachstumsgeschwindigkeit zu einer Blockade der Hirnflüssigkeit im Ventrikelsystem und somit zu einem Verschlusshydrozephalus, dem sogenannten Hydrocephalus occlusus, führen. Daraus resultiert eine Druckerhöhung im Ventrikelsystem, die zu Kopfschmerzen, Übelkeit, Erbrechen, Gangunsicherheit und Bewusstseinseintrübung bis hin zum Koma führen kann.
Ependymome können ausserdem durch den Befall von Hirnnerven zu Ausfällen der Gesichts-, Schlund- und Augenmuskulatur führen.
Bei Säuglingen kann sich ein Ependymom aufgrund der nicht verschlossenen Schädelnähte in einer Vergrösserung des Schädelumfangs (Makrozephalie) sowie einer vorgewölbten und gespannten Fontanelle manifestieren. Dies tritt häufig auf, bevor das Kind andere Symptome wie Entwicklungsverzögerungen, Übelkeit oder Erbrechen zeigt.
Bei intramedullär (im Rückenmark) gelegenen Ependymomen klagen die Patienten in erster Linie über lokale, nicht ausstrahlende Rückenschmerzen. Des Weiteren haben etwa die Hälfte der Patienten zum Zeitpunkt der Diagnose Sensibilitätsstörungen sowie Kraftdefizite.
Eine spinale Bewegungsstörung (Ataxie) bei Tumorwachstum im Hals- und Nackenbereich sowie eine Harnblasen- oder Mastdarmfunktionsstörung treten hingegen selten auf.
Wie wird ein Ependymom diagnostiziert?
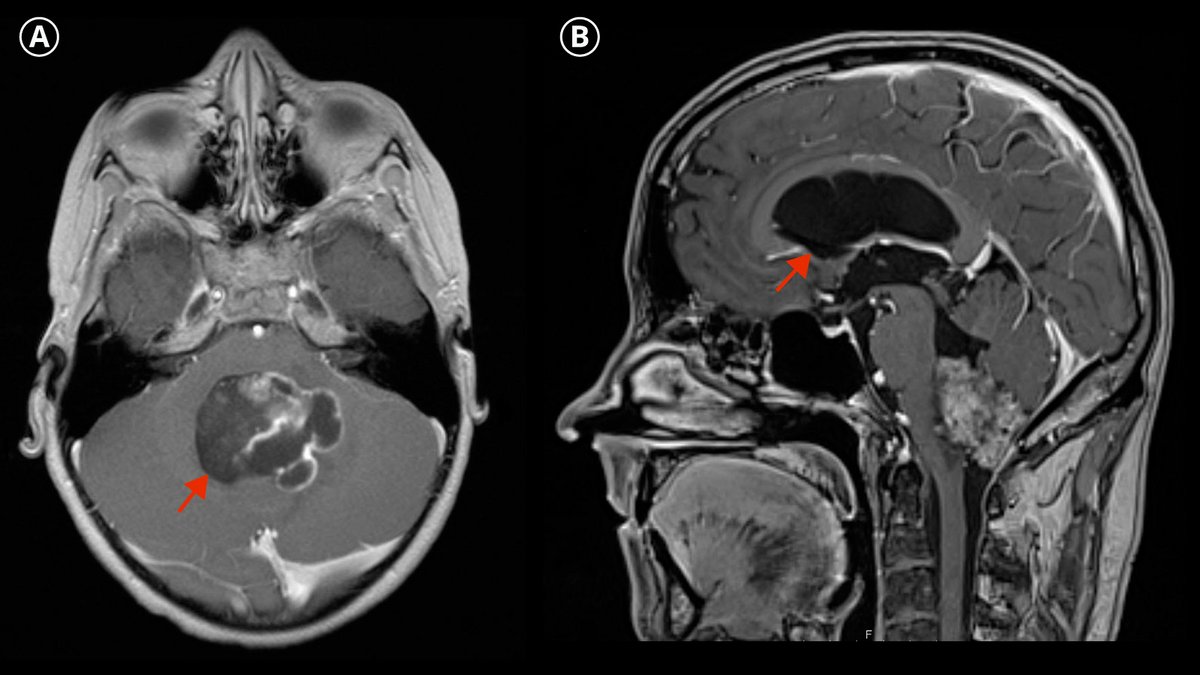
In der bildgebenden Diagnostik zeigen sich Ependymome typischerweise als heterogene Raumforderung mit soliden oder auch nekrotischen oder zystischen Anteilen, wobei Einblutungen und Kalkablagerungen nicht selten sind.
Die bildgebende Diagnose erfolgt durch eine Magnetresonanztomografie (MRT oder MRI von engl. Magnetic Resonance Imaging).
Zum Ausschluss von möglichen Metastasen muss stets die bildgebende Untersuchung des gesamten Zentralnervensystems inklusive des Rückenmarks erfolgen.
Bei bis zu 10 % der Patienten kann es zu einer Aussaat von Tumorzellen in das Hirnwassersystem (Ventrikelsystem) kommen. Man spricht hier von sogenannten Abtropfmetastasen. Darum muss zusätzlich eine Lumbalpunktion zum zytologischen Nachweis von Tumorzellen durchgeführt werden.
Wie wird ein Ependymom behandelt?
Die Therapie von Ependymomen hängt von verschiedenen Faktoren ab, einschliesslich des Alters des Patienten, der Tumorlokalisation, des Tumorgrads und der molekularen Eigenschaften. Im Allgemeinen umfasst die Behandlung folgende Schritte *, *:
Chirurgische Resektion
Die maximal sichere chirurgische Entfernung des Tumors ist das Hauptziel der Behandlung und hat einen starken Einfluss auf die Prognose. Bei speziellen Subgruppen der Ependymome (PFB und YAP-Fusion, spinale Ependymome WHO-Grad 2) kann sogar aufgrund der guten Prognose eine Resektion ohne Strahlentherapie möglich sein. Bei Rezidiven sollte eine erneute Resektion diskutiert werden.
Kann ein einwandfreier Liquorabfluss durch eine maximale und sichere Tumorresektion nicht wiederhergestellt werden, so bedarf es einer zusätzlichen Operation für eine alternative Ableitung des Hirnwassers. Dies kann beispielsweise mittels endoskopischer Drittventrikulostomie oder einem ventrikuloperitonealen Shunt erreicht werden.
Strahlentherapie
Bei den meisten Ependymomen ist eine adjuvante Strahlentherapie erforderlich um das Rückfallrisiko zu verringern. Bei bereits metastasiertem Tumor ist eine intensivere Dosis der Bestrahlung notwendig. Eine Strahlentherapie ist allerdings aufgrund des jungen Patientenalters nicht immer möglich. Deshalb kann insbesondere bei Kindern unter 12 Monaten alternativ eine Chemotherapie eingesetzt werden.
Chemotherapie
Die Rolle der Chemotherapie bei Ependymomen ist umstritten. In einigen Fällen, wie beispielsweise bei zu jungem Patientenalter für eine Strahlentherapie, kann sie als Verzögerungsschritt eingesetzt werden.
Langfristige Nachsorge
Aufgrund des Risikos von späten Rückfällen sowie Langzeitfolgen der Therapie ist eine langfristige Nachsorge wichtig, um ein erneutes Wachstum des Tumors frühzeitig zu erkennen und zu behandeln.
Die spezifische Behandlungsstrategie wird individuell für jeden Patienten festgelegt und kann je nach den individuellen Merkmalen des Tumors und dem Ansprechen auf die Therapie variieren. Für eine optimale Behandlung ist eine interdisziplinäre Betreuung unverzichtbar.
Wie ist die Komplikationsrate bei der Therapie?
Ganz allgemein gilt, dass die Komplikationsrate sowohl der Tumoroperation als auch einer Radiotherapie durch viele Faktoren beeinflusst wird. Grösse und Lage des Tumors, dessen Zugänglichkeit sowie der klinische Zustand und das Alter des Patienten sind hier hervorzuheben.
Je nach Lage des Tumors besteht für benachbarte Strukturen ein gewisses Risiko, bei einer Operation beschädigt zu werden. Zu nennen ist hier vor allem das Posterior-Fossa-Syndrom mit zerebellärem Mutismus nach infratentorieller Operation, was deutlich häufiger bei Kindern vorkommt. Dieses Syndrom führt zum Verlust der Sprache häufig in Kombination mit weiteren Symptomen, wie z. B. emotionaler Labilität, verminderter Muskelspannung und Bewegungsstörungen.
Ausserdem besteht während der Operation das Risiko Tumorzellen zu verschleppen, sodass Abtropfmetastasen entstehen können.
Um den Tumor vollständig entfernen zu können und das Risiko für Komplikationen so gering wie möglich zu halten, verwenden wir am Inselspital spezielle innovative Verfahren wie unter anderem die intraoperative Bildgebung mit MRI, Tumorvisualisierung mit Fluoreszenzfarbstoffen, die Neuronavigation sowie ein spezialisiertes intraoperatives Neuromonitoring.
Wie ist die Prognose bei einem Ependymom?
Die Prognose hängt von verschiedenen Faktoren ab wie der Tumorlokalisation, dem Patientenalter, der Tumor-Untergruppe und dem Ausmass der Tumorentfernung.
Neben der histologischen Gradierung nach der WHO-Klassifikation ist die komplette chirurgische Entfernung der wichtigste prognostische Faktor.
Wichtig ist ausserdem der molekulare Subtyp des Tumors Die Prognose variiert sehr stark innerhalb der verschiedenen Subtypen. Zum Beispiel zeigen supratentorielle Ependymome mit ZFTA-Fusion und YAP-Fusion einen guten Verlauf. Bei den infratentoriellen Ependymomen kommen PFA-Ependymome meist bei jüngeren Kindern vor und sind mit einer schlechten Prognose verbunden. PFB-Ependymome sind häufiger bei älteren Kindern und Erwachsenen und zeigen eine günstigere Prognose mit alleiniger Heilung durch eine chirurgische Entfernung.
Die 5-Jahres-Überlebensrate bei Kindern variiert daher zwischen 50 % und 75 %, wobei die Mortalität deutlich erhöht ist, wenn es sich bei dem Tumor bereits um ein Rezidiv handelt. Durch eine unterstützende Radiotherapie kann die Rezidivrate vor allem bei inkompletter Tumorentfernung gesenkt werden.
Referenzen
-
Lutz K, Jünger ST, Messing-Jünger M. Essential Management of Pediatric Brain Tumors. Children (Basel). 2022 Apr 2;9(4):498. doi: 10.3390/children9040498.
-
Central Nervous System Tumours, WHO Classification of Tumours, 5th Edition, Volume 6, 2021, ISBN-13-978-92-832-4508-7
-
Rudà R, Reifenberger G, Frappaz D, Pfister SM, Laprie A, Santarius T, Roth P, Tonn JC, Soffietti R, Weller M, Moyal EC. EANO guidelines for the diagnosis and treatment of ependymal tumors. Neuro Oncol. 2018 Mar 27;20(4):445-456. doi: 10.1093/neuonc/nox166.
Weiterführende Literatur
- Berlit P. Basiswissen Neurologie. 6. Auflage. Berlin Heidelberg: Springer-Verlag; 2014.
- Chamberlain M. Intracranial ependymoma. In: International Neurology. 2nd Edition. Chichester: John Wiley & Sons, Ltd; 2016.
- Hacke W. Neurologie. 14. Auflage. Berlin Heidelberg: Springer-Verlag; 2016.
- Massimino M, Barretta F, Modena P, Witt H, Minasi S, Pfister SM, Pajtler KW, Antonelli M, Gandola L, Luisa Garrè M, Bertin D, Mastronuzzi A, Mascarin M, Quaglietta L, Viscardi E, Sardi I, Ruggiero A, Pollo B, Buccoliero A, Boschetti L, Schiavello E, Chiapparini L, Erbetta A, Morra I, Gessi M, Donofrio V, Patriarca C, Giangaspero F, Johann P, Buttarelli FR. Second series by the Italian Association of Pediatric Hematology and Oncology of children and adolescents with intracranial ependymoma: an integrated molecular and clinical characterization with a long-term follow-up. Neuro Oncol. 2021 May 5;23(5):848-857. doi: 10.1093/neuonc/noaa257.
- Merchant TE, Bendel AE, Sabin ND, Burger PC, Shaw DW, Chang E, Wu S, Zhou T, Eisenstat DD, Foreman NK, Fuller CE, Anderson ET, Hukin J, Lau CC, Pollack IF, Laningham FH, Lustig RH, Armstrong FD, Handler MH, Williams-Hughes C, Kessel S, Kocak M, Ellison DW, Ramaswamy V. Conformal Radiation Therapy for Pediatric Ependymoma, Chemotherapy for Incompletely Resected Ependymoma, and Observation for Completely Resected, Supratentorial Ependymoma. J Clin Oncol. 2019 Apr 20;37(12):974-983. doi: 10.1200/JCO.18.01765.
- Merchant TE, Li C, Xiong X, Kun LE, Boop FA, Sanford RA. Conformal radiotherapy after surgery for paediatric ependymoma: a prospective study. Lancet Oncol. 2009 Mar;10(3):258-66. doi: 10.1016/s1470-2045(08)70342-5
- Upadhyaya SA, Robinson GW, Onar-Thomas A, Orr BA, Billups CA, Bowers DC, Bendel AE, Hassall T, Crawford JR, Partap S, Fisher PG, Tatevossian RG, Seah T, Qaddoumi IA, Vinitsky A, Armstrong GT, Sabin ND, Tinkle CL, Klimo P, Indelicato DJ, Boop FA, Merchant TE, Ellison DW, Gajjar A. Molecular grouping and outcomes of young children with newly diagnosed ependymoma treated on the multi-institutional SJYC07 trial. Neuro Oncol. 2019 Oct 9;21(10):1319-1330. doi: 10.1093/neuonc/noz069.
- Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F, Wani K, Tatevossian R, Punchihewa C, Johann P, Reimand J, Warnatz HJ, Ryzhova M, Mack S, Ramaswamy V, Capper D, Schweizer L, Sieber L, Wittmann A, Huang Z, van Sluis P, Volckmann R, Koster J, Versteeg R, Fults D, Toledano H, Avigad S, Hoffman LM, Donson AM, Foreman N, Hewer E, Zitterbart K, Gilbert M, Armstrong TS, Gupta N, Allen JC, Karajannis MA, Zagzag D, Hasselblatt M, Kulozik AE, Witt O, Collins VP, von Hoff K, Rutkowski S, Pietsch T, Bader G, Yaspo ML, von Deimling A, Lichter P, Taylor MD, Gilbertson R, Ellison DW, Aldape K, Korshunov A, Kool M, Pfister SM. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell. 2015 May 11;27(5):728-43. doi: 10.1016/j.ccell.2015.04.002.
- Elsamadicy AA, Koo AB, David WB, Lee V, Zogg CK, Kundishora AJ, Hong CS, DeSpenza T, Reeves BC, Kahle KT, DiLuna M. Comparison of epidemiology, treatments, and outcomes in pediatric versus adult ependymoma. Neurooncol Adv. 2020 Feb 21;2(1):vdaa019. doi: 10.1093/noajnl/vdaa019.
- McGuire CS, Sainani KL, Fisher PG. Incidence patterns for ependymoma: a surveillance, epidemiology, and end results study. J Neurosurg. 2009 Apr;110(4):725-9. doi: 10.3171/2008.9.JNS08117.
- Gerstner ER, Pajtler KW. Ependymoma. Semin Neurol. 2018 Feb;38(1):104-111. doi: 10.1055/s-0038-1636503.
- Taylor MD, Poppleton H, Fuller C, Su X, Liu Y, Jensen P, Magdaleno S, Dalton J, Calabrese C, Board J, Macdonald T, Rutka J, Guha A, Gajjar A, Curran T, Gilbertson RJ. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell. 2005 Oct;8(4):323-35. doi: 10.1016/j.ccr.2005.09.001.